
Blood Banking and Transfusion Medicine
Allogenic Donor Testing
- H&P
- Blood collection
- Testing of Donor Blood
Blood Group Antigens
- ABO, Rh, Kidd, Lewis, Duffy, MNS, Kell, Lutheran, P
- Human Leukocyte Antigen (HLA)
Crossmatch
Red Cell Panel
Autoantibodies
Special circumstances
- Neonatal / intrauterine transfusion
- Maternal ITP
- Neonatal Alloimmune Thrombocytopenic Purpura (NATP)
- Hemolytic Disease of the Newborn
- Sickle Cell Disease
- Massive Transfusion
Blood Products
Whole blood
Packed Red Blood Cells (pRBCs)
Platelets (random donor)
Platelets (pheresis)
Fresh Frozen Plasma (FFP)
Cryoprecipitate
Granulocyte concentrate (pheresis)
Transfusion Associated Infections
Transfusiton Reactions
Transfusion-Related Acute Lung Injury (TRALI)
Possible TRALI (pTRALI) / transfused ARDS
Hemolytic Transfusion Reactions (HTRs)
Transfusion-assoc Circulatory Overload (TACO)
Septic Transfusion Reactions
Anaphylactic Transfusion Reactions
Post-Transfusion Purpura (PTP)
Transfusion-assoc Graft Vs Host Dz (TAGVHD)
Febrile Nonhemolyic Transfusion Reactions
Mild Allergic Reactions
Acute Hypotensive Reactions
Blood Banking and Transfusion Medicine
Allogenic Donor Testing
Must give prospective donor questions about medical and social history
- minimum age: 16 years
- donor should report becoming HIV+ w/in 1 year after donating
Testing Principles
2 stages: (1) Sensitization / coating; (2) Bridge formation
(1) Sensitization / coating is the binding of ab to RBC surface and depends on multiple factors
- sensitization is a simple reaction: ag + ab <-> ag-ab
- Ko = equilibrium constant of rxn, where a larger Ko means push to the right side of eqn c more stable and rapid rxn
Affinity of abs and ags depend on several factors:
- cold (IgM) vs warm (IgG) reactive
-- must react in appropriate temp; cold abs usually vs carbohydrate ags (ABO, Lewis, I/i, P, M, N) where warm are usually vs protein ags (Rh, Kell, Kidd, Duffy), and warm abs usually most significant
- size: RBC is much bigger than an ab, which means centrifugation necessary in vitro
- electrical repellence: RBCs have neg charge from sialic acid on surface, and repel each other (zeta potential); to reduce zeta potential can use LISS or albumin (less ions around RBCs) or water-exclusion (PEG); zeta potential is a reason IgG molecules have hard time directly agglutinating RBCs
- pH: optimal pH makes an envt where RBC surfaces negatively charged and abs weakly pos; decreasing pH causes dissociation of ab from RBC surface
- amts of ag and ab: typically use 1 drop RBC and 2 drops of serum, giving a mild ab excess to promote shift of eqn to right; too much ab excess cause prozone effect (inhibits agglutination) and too much ag causes postzone effect (also inhibits agglutination)
(2) Bridge formation is linking of RBCs coated c ab
- IgM more capable of forming bridges bwt adjacent cells 2/2 pentameric structure (10 possible bridging sites) vs IgG which has a harder time 2/2 only having 2 binding regions
- ags that extend from RBC surface, like M, N and ABO make it easier for IgG to directly bind and form bridges, while Rh and other ags closer to the cell surface not usually agglutinated by IgG abs
H&P
Basically just looking at vital signs, hemoglobin and hematocrit, as well as making sure there are no obvious signs of IVDA
- should be fine to donate if within normal limits
- if pt afebrile, there is no deferral period for donors following toxoid admin, synthetic/killed vaccines, bacterial/rickettsial/hep B vaccines
- deferral of 2-4 weeks after receipt of live attenuated
- deferral period 1 yr if in jail > 3 days
- defer 7 days after last warfarin dose, 2 wks after last clopidogrel / ticlopidine dose
- 12 mo deferral after receiving blood product transfusion
- double RBC donations can give after 16 wks
- 12 mo deferral for malaria areas
- 1 mo deferral after last dose of acutane
- 1 yr deferral after needle stick
- indefinite deferral for donors who get viral hepatitis after 11th birthday
- indefinite deferrals for high risk HIV behavior (IVDA, exposure), Receiving money/drugs for sex; Testing positive for HBV, HCV, HIV, HTLV; Receiving clotting factor concentrates (hemophilia); Babesiosis/Chagas’ Disease; Human growth hormone (pre-1985); Bovine growth hormone/insulin; Dura mater graft; Xenotransplantation and etretinate (tegison) teratogen exposure
• Last donation date
> 8 weeks for whole blood
> 16 weeks for DRBC
> 4 weeks for infrequent plasmapheresis
> 48 hrs.. for single apheresis
> 7 days for double/triple apheresis
Blood Collection
Donors give written consent and are notified if any of the lab testing on the blood comes back abnormal
Blood is collected into a closed sterile system
- some of the blood that is left in the tubing can be pinched off by a heat-sealer for further lab testing
A max of 10.5 mL/kg can be collected from someone; a standard unit of blood is 450 mL +/- ~10% (45 mL)
- if a unit has 300-404 mL, it is deemed a "Low volume unit" and cannot be used to generate other blood products (platelets, plasma, etc)
-- if <300 mL, then the amt of anticoagulant in the unit must be lowered appropriately
Donors can have adverse rxn's while donating such as a vasovagal rxn (get bradycardia, should elevate legs and put cold pack on forehead), hypovolemia (get tachycardia, give IV fluids), hyperventilation (have em breath into a paper bag), "citrate effect" in apheresis donors (perioral tingling, arrhythmias, seizures 2/2 hypocalcemia from citrate infusion to donor, give oral Ca2+ to tx), hematoma (apply pressure to site)
Units stored at 20-24 C for first 8 hrs to make other blood products, then red cells stored at 1-6 C
Testing Donor Blood
Forward and reverse typing done for ABO and Rh typing either done on plasma (preferred for gel testing) or on serum
- if donor is Rh neg, testing must be done to test for weak D (D ag variant where pt that is really D-pos tests as D-neg; could expose D-neg pt and cause formation of anti-D; the weak D test is an IAT)
- antibody screen performed on all units to check for ab's to RBC ag's in pts s/p transfusion / pregnancy
- RBCs can be transfused if ab present, but must be labeled c the ab (often get discarded); although plasma and plts not transfused 2/2 large amt of ab
- screening for HbsAg, anti-HBc, anti-HCV, HCV RNA, anti-HTLV-1/-II, anti-HIV-1/-2, HIV RNA, RPR and WNV
- risk of getting HBV from transfusion: 1:800k-1:million
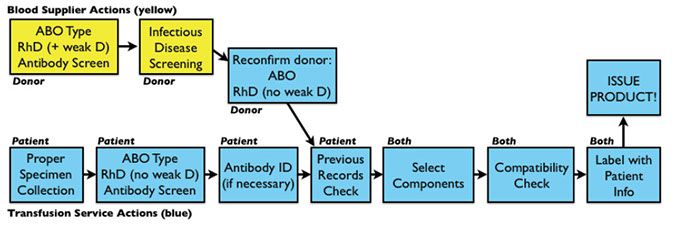
Transfusion service only responsible for:
-Forward grouping of ABO
- only DAT for RhD type, and no weak D if neg
- no req to repeat infx dz screening
Clerical error is MCC fatal transfusion rxn
- need at least 2 unique identifiers per sample
- hemolyzed samples can interfere with tube testing
- blood sample is kept for 7 days post transfusion
Visual inspection of the unit must be done, and unit should be discarded if contaminated (looks purplish), if visibly lipemic, or if hemolyzed or clotted
Labelling occurs after donation record have been compared to previous transfusions, all holds are taken care of, infx dz results are complete (and negative), all quality processes have been complete
Testing Recipient Blood
In obtaining pt sample, ID and labeling critical
- 1/2000 samples has Wrong Blood In Tube (WBIT)
- to collect specimen: (1) request generated, (2) ID pt by wristband and ask their name (if possible); (3) compare wristband and stated ID info to each other and the requisition; (4) draw sample into unlabeled tubes; (5) label tubes at pt bedside (needs at least 2 identifiers; should not prelabel tubes)
Transfusion service compares ID on tube to requisition (must match exactly, absolutely NO errors or changes!); make sure the req has the req'd info
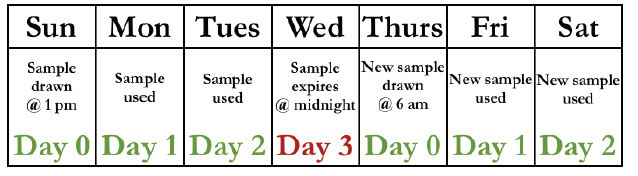
- timing: if pt transfused or preg in last 3 mo, or if hx unknown, blood sample considered predictive (blood at higher risk of developing abs) and a new sample req'd every 3 days (calculated in strange way where day of draw considered day 0 and then expires at midnight on day 3 [see chart])
-- if pt NOT transfused or preg in last 3 mo, sample has no upper time limit (can be used for 30-45 days)
- all samples should be kept at least 7 days after transfusion
-- sample integrity: most labs doing tube testing prefer serum (clotted) while tubeless testing (gel, solid phase) prefer plasma 2/2 clotted debris from incomplete clotting
-- plasma samples can weaken detection of certain abs that are complement dependent (ie Kidd) 2/2 Ca2+ inhib of complement fixation
- hemolysis in plasma makes it impossible to r/o an in vitro hemolytic ab
- lipemia interferes c detection of hemolysis and can interfere c automated testing
Patient samples must get forward and reverse ABO typing, RhD typing by direct agglutination (should avoid weak D test for D-neg pts on direct test) and an ab detection screen
- previous records should be checked to make sure they match (if no prev sample, can redraw or test again on same sample to verify)
Forward typing done to test for ABO and Rh as in donor, and reverse done for ABO
- do not need to check for weak D
-- partial D pts should get D-neg blood so they do not form an anti-D, and most current formulations will call partial D patients D-neg and weak D pts D-pos (which is what you want)
- if pt has been preggers or transfused in last 3 months, need to draw tested sample w/in 3 days of transfusion
- Ab detection ("screen") done by testing pt serum against RBCs from 2-4 fully phenotyped grp O individuals, which is read at AHG (no 37C or IS)
-- must check for D, C, c, E, e, Fya, Jka, Jkb, K, k, Lea, Leb, M, N, P1, S, s
-- if screen is positive, must id the ab, and if the ag is significant, must provide ag-neg blood
-- definition of "significant" ab varies by facility, but most are similar
- must check previous records b4 transfusion (AABB Standard 5.13.5), such as the ABO/Rh type (if no previous must repeat test either on same sample or new draw), hx of clinically significant abs (can disappear over time, but must ALWAYS be honored), prev rxns, special interventions, difficult testing or grouping
Donor and Patient Evaluation
Select products with best chance of maximum benefit and minimum harm based on provider order, blood bank serologic testing, and check of historical records
- ABO compatibility seen in table at right (Modified from AABB Technical Manual, 17th ed, 2011, Table
15-5, pg 447)
RhD compatibility
- with RBC products D-negative premenopausal females should receive D-negative RBCs,
granulocytes, or whole blood unless in dire circumstances
- D-negative males and older females may receive D-positive RBCs when necessary (trauma, massive transfusion, transplant) unless have anti-D
-- In hospitalized patients, risk of anti-D formation in this setting is ~22%
- with Platelet products, same general rules as above, though risk is considerably less than RBCs
(~4% of D-negative patients receiving D-positive platelet products form anti-D)
-- Reasonable strategy: Consider use of prophylactic RhIG to prevent immunization when giving D+ platelets to D- premenopausal females
- Plasma products (FFP/FP24/CRYO) it is not necessary to match for RhD
Antigen-negative RBC components
Required when current or historical testing shows one or more significant RBC antibody
Serologic methods
Units selected by testing with licensed specific antisera (e.g., anti-K, anti-C, anti-Fya, etc)
- may just be for confirmation, as most blood centers have already performed RBC phenotyping on many of their donors
- May be more difficult if just pulling random units off the shelf in a hospital transfusion service
Calculation: Estimated units to screen to find particular antigen profile:
- QUESTION: A donor has anti-K and anti-Fya. How many units should a transfusion service expect to screen in order to find two compatible units?
- ANSWER: Take percentages of antigen negative donors and multiply
(1) Example: K-negative 91%, Fya negative 32%
- Note that these percentages assume a primarily caucasian donor base; adjust according to local situation
(2) 0.91 x 0.32 = 0.29 (29% of donors would be expected to be compatible)
- This is only an estimate, of course
- ABO and RhD status will influence likelihood of finding compatible units
- Divide the number of units needed by the percentage of compatible donors to find estimated units to screen
(1) Example: 2 / 0.29 = 6.9 units screened to find 2 compatible (fairly likely chance of success)
-- A HUGE number, however, suggests the need to call blood supplier and find uncommon/rare units
Molecular methods
(1) Genotyping technology available for screening via single nucleotide polymorphism for genes for an enormous number of antigens
(2) Used in blood centers and transfusion services
(3) Results must still be confirmed serologically when licensed antisera is available
(4) Also useful for determining true genotype of recently transfused patients
Check for compatibility
“Crossmatch” used to determine compatibility between donor and patient
- “crossmatch” usually means “Major” crossmatch, showing compatibility between recipient serum and donor RBCs (vs minor crossmatch which is donor serum vs recipient RBCs)
- the MAIN reason to do a crossmatch is to ensure ABO compatibility!
a) Added benefit: May detect antibody vs. low-incidence antigen not present on screening cells but present on donor cells
b) Also helps detect incompatibility when antibody screen performed incorrectly
- Required before transfusion of any product that contains at least 2 mL of RBCs
(1) Whole blood, (2) pRBCs, (3) Granulocyte concentrate
-- crossmatch NOT needed for transfusion of:
(1) Plasma (FFP or FP24), (2) Platelets (unless heavily contaminated with RBCs), (3) Cryoprecipitate
3 main types of major crossmatch
Serologic crossmatches
1) "Full" AHG crossmatch
(a) Transfusion services may choose to perform AHG crossmatches on all samples, but such a strategy is overkill with no antibody on the screen
- AHG crossmatch is required, however, when patient has history of clinically significant RBC antibodies or has one or more currently
(b) Most commonly uses washed donor cells in 2-5% suspension mixed with patient serum in a test tube with LISS enhancement
- Can use solid phase or gel technology for crossmatch, but it may require additional steps to prove ABO compatibility
- LISS/gel > PEG/albumin/saline > solid phase
(c) The only phase that MUST be read is AHG, however, agglutination or hemolysis after 37 C incubation is also a positive reaction showing incompatibility
2) Immediate-spin (abbreviated) crossmatch
(a) By definition, may ONLY be performed if antibody screen is negative and there is no history of significant RBC antibodies
(b) Is simply a final ABO compatibility check
(c) Procedure:
i) Mix patient serum with donor 2-5% RBC solution (2 drops serum to 1 drop RBCs)
ii) Centrifuge and observe for agglutination or hemolysis
(d) Why do it?
i) Saves time and reagents
ii) Decreases workload for transfusion service workers
iii) Demonstrated to be safe (<0.1% risk of acute hemolysis); this rate is actually very similar to that with an AHG crossmatch
3) Electronic (“computer”) crossmatch
(1) Like immediate-spin crossmatch, may only be used when current antibody screen is negative and there is no history of significant RBC antibodies
(2) Other requirements:
(a) FDA-approved, locally validated computer system capable of making logic judgments about ABO compatibility between donor and patient
i) Part of the validation includes demonstrating that the computer will ALERT the transfusion service when it sees incompatibilities
(b) Patient who has had two separate ABO determinations (including one for this transfusion episode)
i) Acceptable: Historical ABO type and current
sample ABO type
ii) Acceptable: No historical ABO type, test current sample ABO type twice
iii) Acceptable: No historical ABO type, test current sample ABO type, require a second ABO type from
a second phlebotomy
(c) Why do it?
i) Potential to save LOTS of time (even more than immediate spin)
ii) Decreased workload and reagent cost in the transfusion service
iii) No significant difference in safety compared to immediate spin or AHG crossmatch (same less than 0.1% risk of hemolysis)
Issues with positive crossmatch results
a) Positive crossmatch after negative antibody screen
1) Positive immediate-spin crossmatch
(a) Donor RBCs are ABO incompatible with recipient antibodies
(b) Anti-A1 in a group A2 or other A subgroup pt
(c) Cold-reactive antibodies in recipient tested only for warm antibodies
(d) Polyagglutinable donor RBCs
2) Positive AHG crossmatch
(a) Antibody vs. low-frequency antigen on donor RBCs
(b) False negative antibody screen
(c) Donor RBCs coated with antibody or complement (positive DAT)
b) Positive crossmatch after positive antibody screen
(1) Autocontrol positive
(a) Warm autoantibody
(b) Antibody vs. recently transfused RBC antigens
(c) Cold autoantibody
(d) Passive alloantibodies (IVIG, transfusion, transplantation, RhIG)
(2) Autocontrol negative
(a) Expected with antibody vs. high frequency antigen
(b) If unit selected as antigen-negative:
i) Incorrectly performed antigen testing
ii) Incorrectly identified antibody
iii) Antibody vs. low-frequency antigen on donor RBCs
Label components
1. Component must have a tag or label affixed that includes:
a) The recipient’s two independent identifiers
b) The donor unit number
c) Results of compatibility testing (if performed)
2. Other information will already be on the standard label, including (to name a few):
a) Component name
b) ABO/RhD type
c) Expiration date and storage temperature
d) Collection facility
e) Approximate volume
Final clerical checks
1. At issue:
a) Verification of patient records noted above as well as component characteristics; the request, component, and records all must match
b) The following are required by AABB Standards (27th ed):
Recipient information:
(a) Two independent identifiers
(b) ABO group
(c) RhD type
Donor/product information:
(a) Donor identification number
(b) ABO group
(c) RhD type (if required)
(d) Compatibility testing results (if applicable)
(e) Special requirements (irradiation, leukocyte reduction, washing, etc.)
(f) Expiration date/time
(g) Issue date/time
c) Check of all of the above is usually done with person checking the blood out of the transfusion service, and must be documented
2. At bedside
a) Usually out of transfusion service control, but is VITAL!
b) Pre-transfusion verification required by AABB Standards (27th ed.); which is the same list as above except for checking issue date/time:
c) This is really the “last defense” against mistransfusion, and transfusing staff must be thoroughly trained and aware of importance of this final check
Testing/ordering nomenclature
A. Hold clot
- Uncommonly used
- Clotted sample held in transfusion service but is not tested at all
B. Type and hold
1. Uncommonly used
2. ABO and RhD typing done, but no other testing (no antibody detection)
C. Type and screen
1. Should be MC pretransfusion order
2. Check of previous records for comparison with current results only
3. ABO, RhD typing done, antibody detection performed
a) If antibody present, identification is performed
4. Very simple to convert from a type and screen to a type and cross, if necessary
a) If antibody screen is negative, only an ABO check is required (accomplished via immediate spin or computer crossmatch)
b) If antibody screen is positive, most transfusion services automatically identify antibody and convert test to “type and crossmatch” below, after selecting
antigen-negative donor RBCs (if antibody is clinically significant)
D. Type and crossmatch (“type and cross”)
1. Same as type and screen, but adds crossmatching (serologic or electronic) for a specified number of units of RBCs
2. RBC units are then designated (reversibly) for that patient
3. Effective strategy: Maximize T&S, minimize T&C whenever possible
E. MSBOS
1. “Maximum Surgical Blood Ordering Schedule”
2. Hospital-specific guide to appropriate routine ordering quantities for specific procedures
3. Generally, a list of surgical and other procedures followed by a recommended blood order for that procedure
a) Order may be “None,” “Type and Screen,” or “Type and Crossmatch for (X) Units”
4. Helps conserve resources and promotes consistency
5. Must be formally approved (with maximum physician input) and promoted widely
to be effective
6. Not required (except in certain states), not widely utilized effectively



ANY significant antibody triggers serologic crossmatch


Blood Group Antigens
In addition to testing, check pt record to see what results for ABO, Rh, and alloantibodies were previously found
- some alloantibodies (Kidd) become undetectable over time but still are significant!!
Blood group antigens are protein, glycoprotein, or
glycolipid on RBCs, detected by an alloantibody
- NOTE: Antigens are not limited to RBCs
Blood group system: Group of blood group antigens
that are genetically linked (30 total systems per ISBT)
- Significance: “Significant” = antibody causes HTRs or HDFN; most significant antibodies are “warm reactive”; meaning they react best at IAT (37 C).
- Most insignificant antibodies are “cold reactive”;
meaning they react best below 37 C.
- Warm antibodies most often IgG, colds usually IgM.
- IgM antibodies are usually “naturally occurring” (no
transfusion or pregnancy required for their formation).
- ABO is the exception
ABO and H systems
2 types of precursor polysaccharides:
- type 1 mostly glycoproteins in secretions (except CSF) and plasma (serum) carrying free-floating proteins (***flies solo [1]***)
- type 2 mostly glycolipids carrying bound ags on RBC surface (*** type 2 is paired, to RBCs or other stuff***)
- if unbranched, called i ag; branched are I ag
-- i ag predominates in neonates; branching inc c age
- modifications to type 1 precursors in serum responsible for Lewis group (see Lewis below)
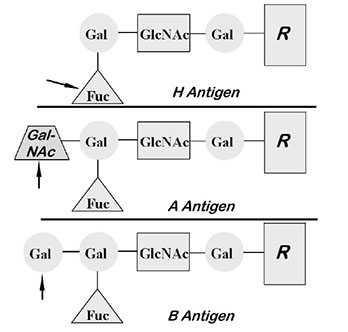
- H ag req'd before A and/or B can be made on RBCs (type 2 H) or in sections (type 1 H)
Se gene (FUT2; where FUT = FUcosylTransferase) is a secretor gene on cr 19 that makes precursor to A or B ags in secretions
- FUT enzyme adds fucose to type 1 chains at terminal galactose, where the product is type 1 H ag; there is an 80% gene freq
- Se = active gene, se = non-active (most [80%] have at least 1 active gene; 20% are sese, and non-secretors)
- makes H ag, except on diff chain
H gene (FUT1) is closely linked to Se on cr 19 and the FUT enzyme product adds fucose to ONLY type 2 precursors on terminal galactose on RBC surface; making the type 2 H antigen; there is 100% gene freq (Bombay = hh is very rare)
- further modifications can make it A or B; no further modifications will leave it as O (see ABO below)
-- thus, as more A or B is made, less H remains
--- H amt: O > A2 > B > A2B > A1 > A1B
Testing done with forward (cell) typing (antisera A and B added to pts blood to see which antigens on RBC surface membrane) and reverse (serum) typing (A and B test cells added to pt serum to see which ab's present)
*** forward on the cell, reverse in the sera***
- strength of rxn graded 0 to 4+ and mf+ (if 2 pops of cells present, some react and some don't)
3 possible ab's: anti-A, Anti-B or anti-AB
- in grp A or B, anti-B or -A are usually IgM, but reacts strongly at body temps; however, grp O anti-A and -B are mostly IgG and react strongly at body temp
ABO discrepancies can arise bwt forward and reverse rxns, which can be 2/2 ag/ab probs or technical error
- ag probs include missing ags (A or B subgroups, transfusion, leukemia), unexpected ags (acquired B, polyagglutination, recent BMT) or ab probs (missing abs in immunodef, neonates / elders) or unexprected ab's (cold abs, anti-A1) or technical errors
Cr 9 alleles encode transferase enzymes that allows transfer of sugars onto H ag to form A (N-acetylgalactosamine; NAG) and B (galactosamine) antigen
- O type does not have a specific antigen added to H and is considered non-functional
- ABO ags on fetal RBCs at 6 wks gestation, and reach adult levels by age 4; they are also on plts, endothelium, kidney, heart, lung, GI, pancreas
ABO incompatibility is among the MC BB fatalities (usually caused by clerical errors) from severe acute hemolytic transfusion reactions
- ABO incompatibility is MC HDFN, though is usually mild
H-deficient types
H-deficient non-secretors ("Bombay" or "Oh")
- No H, A, or B 2/2 no H or Se;
Genotype: hh, sese
Strong anti-A, -B and -H; O forward and reverse, screen positive
Multiple rare FUT1 mutations id'd (1:10k in India, 1:1M in Europe); FUT2 mutations much MC (20% or so are sese)
- 1950's in Bombay, India (now Mumbai)
- Grp O, but incompatible c grp O (no rxn against anti0A1 or anti-B)
- grinding up the seeds of Ulex europaus (common plant) that makes H lectin normally reacts against H ag, but would be neg in pt c Bombay phenotype
- testing Oh secretions are neg for A, B, H and LeB bc have no ag's on their surface
- asoc c Leukocyte Adhesion Deficiency, type II (LAD2); assoc bc neutrophils need fucose to attach to endothelial cell surface, presents c MR, infx
- since, has been seen elsewhere, but rare
H-deficient secretors ("Para-Bombay" or "H+W")
Have Se
- hh, SeSe, or Sese (mut in FUT1 allele, but no del in secretor allele)
- have no H on RBC surface (like Bombay), but secretions can have FUC attached
- some (a small amt) of RBCs can have H, which is leaked in from secretions
- in an A1 secretor (AO or AA) can have weak rxn against Anti-A1, no rxn against B, and weak / absent rxn c H-lectin (Ulex europaeus)
Mostly H-deficient people ("H+W")
- weak H (FUT1), sese
- Ah makes sig anti-H and anti-B
- Bh make anti-H and anti-A
Antibodies
Sig ab: anti- H at body temp
Insig abs: anti-H below body temp, and anti-HI common and b9
- both common in A1 and A1B
Tx for H-deficient pts
Both Bombay and Para-Bombay need H-neg blood (from Bombay donors) if they are making anti-H alloab
- highly sig IgM ab
- destroys H-pos RBCs at body temp
- all but other Oh hemolyzed
Cold reacting ("auto") anti-H or -HI very common in those c little H normally
- there abs NOT a prob, cold-reacting IgM, no hemolysis at body temps
In USA: O (45-50%)> A (25-40%)> B (10-20%)> AB (~5%)
- for H ag: O>>A2>B>A2B>A1>A1B
Group O
MC blood group across races
Have OO genotype bc do not inherit A or B genes, and produce IgM anti-A, IgM anti-B, IgG anti-A,B
- bc strong IgG component, mild HDFN common in O moms, though fetus makes ABH to neutralize abs
Group A
Have either AA or AO genotype and make the A transferases to put N-acetylgalactosamine (NAG) on H ag (have A and H ags and make IgM anti-B)
- A subgroups: A1 people (80% of A's) make more A substance (thus have less H ag) than A2 people (20% of A's; have relatively more H ag than the A1s)
- A1 RBCs have ~5x more A on RBC surface than A2
-- anti-A1 in 5% of those c A2 and ~1/3 of those c A2B, which is usually in insignificant IgM, but is a common cause of ABO discrepancies (though should avoid A1 transfusion if reactive at 37C)
-- thus can transfuse type A1 to A2
-- can differentiate A1 from A2 by strength of rxn against B serum or Dolichos biflorus lectin (both of which have anti-A, though will agglutinate A1 but not A2)
Group B
Either BB or BO genotype, and B and H ags with galactose moiety, make IgM anti-A, but no important subgroups (like those A has)
The Acquired B phenotype
- A1 RBC contact c enteric gram neg: Colon ca, intestinal obstruction, gram neg sepsis
- AB forward (weak anti-B rxns); A reverse
- Bacterial enzymes deacetylate group A GalNAc; remaining galactosamine looks like B and reacts c forms of monoclonal anti-B (ES-4 clone)
- use monoclonal anti-B that does NOT recognize acquired B, acidify serum (no rxn c anti-B)
The B(A) phenotype
- Opposite of acquired B (grp B pts c weak A activity); an inherited (not acquired) condition
- cross-rxn c specific monoclonal anti-A; test using different anti-A shows the pt as B
Group AB
Least freq blood type (~1/20) have AB genotype, and make A and B ags (have very little H); may further subdivide into A1B or A2B depending on the A ag
- make no abs
Rh
2nd most important blood group (after ABO)
- the old (incorrect) Rh ag terminology systems included Fisher-Race (DCE or CDE, c 5 major ags [D, C, E, c, e] where "Rh+" really meant "D+") and Wiener (Rh-Hr, which had 5 main ags and archaic names, which are still essential to know although their previously postulated inheritance has been disproven [see chart])
- to convert from Wiener to Fisher-Race terminology: R=D, r=d; 1 (prime) = C, 2 (double prime) = E; 0 (blank) = ce; any sub-/superscript letter = ce
- only R1, R2, R0 and r occur c significant freq
-- to remember: R0 is MC in blacks, least common in white; r is always second in freq, R1 always before R2 (see chart)
Anti-D sera added to blood sample (forward typing) to check for agglutination
- weak-D checked on all D-negative donor blood but not recipient's
- the only pts that definitely need weak D testing are apparently D-neg babies c D-neg moms
- weak D moms do not need RhIG prophylaxis
RHD and RHCE loci on cr 1, which encode Rh antigens; D type determined by presence/absence of RHD
- One protein gives both C/c and E/e ags, the combo determined by which alleles of RHCE present (CE, Ce, cE, or ce)
- RHAG gene on cr 6 codes for Rh associated glycoprotein (RhAG)
- the Rh ags complex with RhAG on the RBC membrane
r/r (cde/cde) is the MC Rh-negative genotype in whites (40% of total Rh) and blacks (30%), while the MC Rh-positive genotype is R1/R1 or R1/r in whites and R0/R0 or R0/r in blacks
Weak D antigen is negative at IS and at 37C with anti-D reagent but positive at AHG with anti-D
- may be 2/2 either a quantitative weak D (MCC, MC in blacks c Dce/Ce making C allele trans to D [Ceppelli effect]) or a partial D (aka "D mosaic; lacks certain epitopes; women c D+ pregs at risk for making anti-D abs (they need RhIG prophylaxis), where abs form against absent parts of RHD; MC is DVI aka D-6 in whites which is a monoclonal anti0usually that usually types these as D-negative)
- weak D can be caused by a mutated form of RHD or RHCe on opposite chromosome to RHD ("C in trans") that inhibits D expression
- if previously sensitized D- people get a weak D, hemolysis may result; though a D- recipient won't be sensitized (form abs) against a weak D donation/transfusion
- partial D vs weak D may be impossible w/o molecular testing; and if in doubt for prenatal testing must consider the pt D-negative
- Del ("D-E-L" pts appear D0neg but have timy amts of D seen after elution of reagent anti0D from RBCs and is primarily in Asian populations (up to 1/3 of D-neg Asians)
- these abs go together: anti-E formation commonly accompanied by anti-c
- think Big 4: R2R2 gives both E and c exposure
Compound Rh ags
- G is an ag present when either C or D present; anti-G reacts against D+C-), D-C+) or D+C+) RBCs (rarely against D-C-G+), commonly presenting as D0neg pt forming anti-D when not obviously exposed to D, and is important bc if D-neg mom has anti-G she still needs RhIG to prevent anti-D; this can cause HTRs (must give D-C- blood)
- f is presnet when RHce is inherited (r and R0); anti-f often seen c anti-e or anti-c and cause mild HDFN or HTR
Rh null peoples have no Rh/RhAG causing RBC stomatocytosis (RBCs leak Na and K, causing hemolytic anemia); and the RBCs also lack LW and Fy5 and have weakened S, S, and U ags
D-negative phenotype
- unusual bc caused by mutations and deletions rather than synthetic actions of a gene product
- in whites have a deletion of RHD gene
- in blacks have point mutations in RHD gene (pseudogene)
- in Asians have inactive RHD gene
Rh antibodies are IgG and only can be acquired by exposure (not like ABO)
- D induces the most abs, then c and E
- very immunogenic: 4/5 D- people exposed to a single unit of D+ develop abs
- all Rh abs except D display dosage
- all Rh ags enhanced by enzymes
- Rh abs can cause extravascular hemolysis c hemolytic dz of newborn
- if anti-E abs found in serum, suspect presence of anti-c bc most pts would have the R1/R1 phenotype (CDe/CDe) after being transfused c R2 (cDE) blood
-- this anti-c is usually undetectable but may cause a delayed hemolyic transfusion reaction (DHTR)
Antibody Testing (Screen/Panel)
Ab Screen done against 2-3 test cells that are O neg to check for alloantibodies, which can cause hemolysis or hemolytic disease of newborn, though not as much as ABO incompatibility
- the screen is an Indirect Antiglobulin Test (IAT), meaning that only the pt serum and AntiHuman Globulin (AHG) added to each test cell
-- an autocontrol group is run simultaneously with the test cells in which pt serum and blood added to AHG
- screen can be done at room temp and 37 C or 37 C only (usually just done at 37 C bc room temp only detects insignificant alloantibodies [anti-M, N, Lewis, I, P])
- if there is any rxn found on the screen, an Ab Panel must be run to see which ab is reacting
- if no rxn found on screen, proceed to crossmatch
Ab Panel is basically just a screen that uses 10 different test cells (all O neg)
- can be done in test tubes or using fancy machines
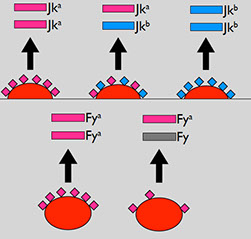
Kidd
Called the "tricky Kidd" bc hard to detect, these warm IgG abs are only produced after an exposure and may disappear over time (for which it is preferred to give Kidd negative blood) and can cause DTH or DHTR, though usually not HDN
- Kidd ags = Jk^a, Jk^b, Jk3 (very high freq, where Jk^a slightly MC than Jk^b in blacks but similar in whites and Asians)
- ags reside on a urea transport protein
- Kidd abs are exposure-requiring, warm-reacting IgG that can fix complement (c IgM component) and cause severe acute HTRs
- displays dosage: homozygotes [Jka+b-)] make more ag than heterozygotes [Jk(a+b+)]; which can give confusing panel results and a false-negative crossmatch
-- also the Kidd does not like to be stored and may be false-neg at a reference lab; also it fades quickly
- usually only reacts at the AHG phase
- most famous for delayed HTRs (anamnestic response, intravascular and often severe)
- mild HDFN at worst (can get if kid only 1 ag different from mom)
23% of US is Jka-, while 28% (60% of blacks) Jkb-
- 72% of whites are Jk(a+,b+) and their antibodies can cause moderate to severe hemolysis
Lewis
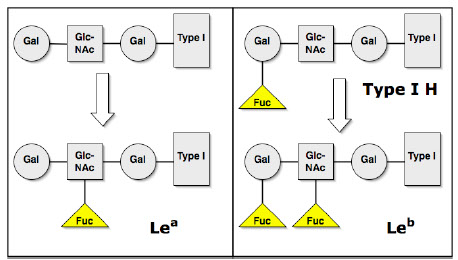
Only type I chains; Le gene makes a fucosyl transferase (FUT3) which puts a fucose on the subterminal GlcNAc type 1 precursors in serum and in secretions, thus making the Le^a ag (le gene is nonfunctional, kinda like h), and then become passively adsorbed onto RBC surface
- not well developed in newborns and not intrinsic to RBCs
Se gene can add another fucose to type 1 precursors only if one is already there (already done by Le gene) making Le^b
- so making an Le^b uses an Le^a, and Le^b can't be made without Le^a and both Le and Se genes (no Le gene mean no Le^a or Le^b)
- ??? although BBGuy says that in secretors the Se product (FUT2) adds fucose then Le product adds fucose making Le^b, and that Le^b is not made from Le^a ???
- non-secretors are always Le^a
- Le (a-b+) still have Le^a, just in very small amts
Lewis abs are insignificant and are made almost only in blacks (~1/4 of blacks) who are Le(a-b-)
- transfused RBCs get the Lewis phenotype of the recipient
- Lewis ags dec during preg
- Le(a-b+) pts do not make anti-Le^a
- H pylori and Norwalk virus attach to gastric mucosa via Le^b ag
- Le(a-b-) pts at risk for Candida and E coli infx
Le can not be accurately typed until 2nd b-day
- most people are Le(a-b+) or Le(a+b-), though nearly 1/4 blacks are Le(a-b-) [nobody is Le(a+b+)???]
I System
Ags built on type 2 chains; expression dependent on age (simple chains on neonates make i ags, branched chains in adults make I ag [Big I in big ppl, little i in little ppl])
- Asian adults more commonly lack I, known as i-adult
- if abs make, are cold-reacting IgM and are insignificant
- Auto-anti-I assoc c cold agglutinin dz and Mycoplasma pneumoniae infx
- Auto-anti-i assoc c infx mononucleosis
- I-adult phenotype assoc c cataracts and HEMPAS
Duffy
Comprised of the antigens Fya and Fyb, which can be destroyed by enzymes
- Fya from Fya gene has high freq in Asians
- Fyb from Fyb gene has high freq in whites
- absence of both ags (Fy (a-b-) is MC Fy phenotype in blacks
- Fy(a+b-) > Fy(a-b+), except in blacks where Fy(a-b-) seen in ~2/3 which makes them resistant to malaria P vivax and knowlesi
Warm IgG antibodies against Duffy antigens are acquired by exposure and show marked dose effect
- Fya abs more common and significant than anti-Fyb
- may cause HTR and severe HDN
MNS
Group comprised of the antigens M and N seen on glycophorin A; while S, s and U on glycophorin B on the RBC surface
- M and N ags seen c equal freq (~3/4); s MC than S (1/2 whites and 1/3 blacks)
- if S-s- (2% blacks) can also be U-negative
- Vicea graminea lectin reacts against N ags
- MurL hybrid ag seen in ~10% chinese
- M and N ags can be destroyed by enzymes
MNS abs display dosage
- M and N abs mostly opposite of S, s, and U abs
- Anti-M and -N are clinically insignificant cold reacting IgM abs that are found naturally, but anti-S, -s, and -U are acquired by exposure and are clinically significant warm-reacting IgG abs
N-like ag ('N')
- GPB always has terminal 5 amino acid seq that matches GPAs terminal sequence when expressing N, known as 'N', which is not really a true N ag, but close enough to precent most M+N- from making anti-N
- seen in all except those that lack glycophorin B; <1% blacks lack S, s and U, and is rare in whites, and anti0N nearly exclusive to blacks
- auto-anti-N induced by hemodialysis by formaldehyde sterilization of machine and modification of N leading to rare auto-ab
Kell
Mrs. Kelleher (1946) has baby c anemia
- direct agglutination test neg
- Dr. Robin found that sometimes abs bind RBCs but dont agglutinate, and thus devised the AHT/IAT/Coomb's test that added component (anti-human globulins) to bind those ags and coagulate
- Kell is short for Kelleher,
- k is antithetical to K
- Kp^a/b/c are Kell-related; or "p" for "Penney"
- Js^a/b for "John Sutter"
Genes: KEL on cr 7 (7q34)
- large gene c 19 exons
- 732 Amino Acid glycoprotein (Kell, CD238)
- up to 18k copies per RBC
- found on BM, testes, and fetal liver
KEL without mutations has reference allele as KEL*02, with multiple high freq ags: K / k Kpa / b / c Jsa/Jsb
Kel with one mutated allele: has all the usual ags with ONE added low freq ag (K, Jsa, Kpa etc)
There are other less common pairs of Kell ags, including KEL 11 / 17, KEL 14 / 24, KEL 31 / 38
K antigen(KEL1, "Big K") - not "Kell" or "K1"
- freq: 9% whites, 2% blacks, rare in Asians, up to 25% in some Persian populations
- expressed in fetal RBCs at 10 WGA
- resistent to proteolytic enzymes (though can get dissolved with Disulfide-breakers (2 mercaptoethanol (2-ME), dithiothreitol (DTT), 2-aminoethyisothiouronium bromide [AET])
k antigen (KEL2, "little k") originally called "Nocella" then "Cellano" is high-freq (99.8-100%), with 2 mutated KEL alleles for K+k-
Anti-K
MC non-ABO after anti-D
- IgG that reacts best at IAT, coats RBCs but does not fix complement, and leads to extravascular hemolysis
- transfusion / preg: 10% efficiency after transfusions
- Less after K+ preg
- rarely "naturally occurring" (assoc c E coli and TB)
- will see indirect bili and spherocytes (from spleen)
Anti-K HDFN
- unique HDFN bc not terribly "hemolytic"
- K ag on RBC precursors (proerythroblast, vs late erythroblast for D)
- anti-K HDFN is suppressive of bone marrow, not hemolytic, c reticulocytopenia and anemia (vs anti-D HDFN c hemolysis, reticulocytosis, hyperbilirubinemia and anemia)
- not freq seen bc K- mom, K+ dad uncommon (4-5% of pregs), and preg not a good immunizer (~80% anti-K from transfusion)
Anti-K and Preg
K- mom c anti-K: consider fetal DNA (mom's serum) or test dad for K (less good); may also consider a titer (not predictive), fetal MCA-PSV, cordocentesis and direct Hgb measurement if concerned
- amniocentesis not useful bc Kell abs suppress erythropoiesis; thus fetal bilirubin conc not reflective of degree of fetal anemia
anti-k
uncommon, if present k- compatible units hard to find
- similar to anti- (extravascular), HDFN (suppressive)
- should also get significant dec when DTT added
Kp antigens
- Kp^a in 2% whites, others rare
- Kp^b nearly 100% in all rares
- Kp^c rare in all races
- all are thiol-sens
Kp antibodies
all are IgG that react best using IAT
- all can cause HTRs and HDFN
- prob of anti-Kp^b: rare, but big prob finding Kp^b-neg compatible donors
- consider banking autologous blood and siblings
Kpa Not Friendly!
the presence of Kpa weakens other Kell system ags, while Kx is slightly increased (usually inconsequential)
Js antigens
- Js^a in 20% of blacks
- Js^b high freq in all
Js antibodies
- analogous to others discussed
- anti-Js^b only seen in Js(a+b-) blacks
- can be big issue in sickle cell pts
- less predictably pathologic
- consider banking autologous blood and testing siblings
Xk protein made by XK gene on cr X, is a transmembrane protein adjacent to Kell glycoprotein
- very important group clinically and seologically
k ag (99.8%, KEL2, Jsb, Kpb) >> K ag (9%, aka KEL1, Jsa, Kpa)
- ags not affected by enzymes, but can be destroyed by ZZAP and DTT
- encoded on cr 7
- Kx ag: bound to Kell glycoprotein on RBC surface and is reqd for proper Kell ag expression and is actually considered a separate blood group (Kx sysem)
- when Kell ags dec, Kx inc; and when Kx dec (as in McLeod sundrome) Kell ags dec as well
- Kell system ags destroyed by thiol reagents but not by enzymes alone
McLeod phenotype:
Dr Allen testing dental students, one dental student had very weak Kell ags across the board, bc had absent Xk protein (from XK del / mutation)
- may have been in King Henry VIII
- no Kx protein produced (found on cr x) which is a supportive protein similar to RhAG
- lack of Kx causes hemolysis and dec RBC survival bc RBCs cannot deform well
- no anti-Ku (as seen in K0), but can form anti-Kx and anti-Km (Kell McLeod), where the pt only compatible c McLeod RBCs
- also assoc c acantholysis, muscular dystrophy, and X-linked Chronic Granulomatous Disease (CGD) when assoc c CYBB deletion
- will be neg for Km protein (since Km protein dependent on Kx-Kell cysteine bonds)
- Tx: avoid transfusion!
Kell null phenotype (K0)
- all Kell ags dec, and Kx inc (probably actually decreased, but since assoc Kell-cloud not there it seems inc from reactions)
- can get significant anti-Ku ("universal") if exposed
- KEL nonsense mutations usually (>20 kniown, all rare)
- anti- Ku - all RBCs incompatible except K0
Kmod
Group of KEL mutations (>10) c single nucleotide polymorphisms, that all lead to markedly dec Kell ag expression
- may require adsorption or elution
- Kmod pts can form anti-Ku-like abs
- neg c self cells
- abs not uniform
- diff mutation = variation
Kell abs are anti-K and are warm IgG and acquired through exposure (transfusion > preg), which can cause HTR and HDN
- anti-k very uncommon 2/2 high ag freq
Lutheran
Lu^b (99.8%) >> Lu^a (5-8%)and are destroyed by enzymes
- Lutheran abs usually not significant and uncommon
P (the cool one)
P1, P and P^k ags are carbohydrate ags (there is no p ag in the P system)
- also built on ABO-related chains
- P and P^k not in the system officially anymore
- MC phenotype is P1 (P+P1+P^k-)
- rarely, pt lacks all three and makes anti-PP1P^k and get acute HTR and early spontaneous abortions
- P ag is the parvovirus B19 receptor
- P6k ag is a receptor for bacteria and toxins
P1 phenotype (16/20 whites, 19/20 blacks): anti-P1+, P+, PP1P^k+, and P^k-
P2 phenotype (4/20 whites, 1/20 blacks): anti-P1 -, P+, PP1P^k +, P^k -
p phenotype: no P ags (anti-P1 -, P-, PP1P^k -, P^k -) though make a powerful anti-PP1P^k which can be broken down by adsorptions into its 3 components, and anti-PP1P^k assoc c DHTR and HDN
Anti-P1 is a cold reacting naturally occurring insignificant IgM
- titers inc c hyatid cyst dz (Echinococcus) and bird handlers (bird feces have P1-like substance)
- anti-P1 can be neutralized by hyatid cyst fluid and pigeon egg whites
- assoc c paroxysmal cold hemoglobinuria bc is a biphasic IgG c anti-P (not P1) specificity, that binds in cold temp and hemolyzes when warmed (aka the Donath-Landsteiner biphasic hemolysin)
- historically in syphilis, now seen post viral infx in kiddos
Diego System
>20 ags built on "band 3", which is important in RBC structure and carries HCO3 anions out of RBCs (for CO2 removal) and anchors membrane to cytoskeleton
Diego ags
Di^a and Di^b are antithetical pair
- Di^a is very low except in South Am and Asians
- Di^b very high in all populations
Wr^a and Wr^b are antithetical pair
- Wr = Wright
- Wr^a is very low freq and Wr^b very high freq
Diego abs
Di abs are IgG while Wr abs have IgM component
Both anti-Di^a and anti-Di^b can cause HDFN that can be severe, but not HTRs
- anti-Di^b can show marked dosage effect
- anti-Wr^a is common, naturally occurring and can cause both HTRs and severe HDN (IgG and IgM)
- anti-Wr^b rarely seen as alloab in AIHA
Abs c "High Titer, Low Avidity" (HTLA) features
- are high freq ags that are generally clinically b9
- Chido, Rogers most freq; which are complement components (C4)
- others are also known (Knops [Kn^a], McCoy [McC^a], and JMH)
- should use caution bc some abs c similar features can be significant (anti-Vel and anti-Yt^a)
Colton (Co) System
Ags (Co^a and Co^b) found on water transport membrane protein (aquaporin 1)
- Co^a very high freq (~100%), Co^b in ~1/10
- both abs can cause significant HDFN
Dombrock System
Do^a/Do^b ags; Do^b more freq
- either ab can cause HTRs but not HDFN and is a warm-reactive IgG
- High freq ags Jo^a, Gy^a and Hy can cause mild HTRs or HDFN, but abs are rare, nearly 100% incidence of these
- Jo^a- and Hy neg exclusively in blacks
- Gy^a neg in Japanese and eastern Europeans
Landsteiner-Weiner (LW) System
LW^a ag more abundant on D+ RBCs
- LW ags originally thought to be Rh ags; but abs not generally significant
Sd^a (Sid) ag
High freq (96%); refractile, small immune complexes c naturally occurring IgM
- lectin of Dolichos biflorus agglutinates Sd^a pos RBC (like A1) that can neutralize c guinea pig or human Sd^a positive urined
Vel Ag
Very high freq ag (>99%); ab is mic of IgG and IgM, and can cause severe HTRs and HDFN, and can interfere c ABO typing 2/2 reaction at room temps; can be allo- or auto-ab
Xg system
- gene on X cr (X-linked) and found in 2/3 males and 9/10 females; abs are insignificant
Yt system (formerly Cartwright)
Yt^a (very high freq (99.8%) and Yt^b (8%); abs are IgG but not usually significant (although anti-Yt^a can cause HTRs)
Human Leukocyte Antigen (HLA)
Major Histocompatibility Complex (MHC) genes on cr 6p encodes HLA and are so close together on 6p that they do not cross when inherited from each parent and are some of the most polymorphic genes in the human genome
- HLA is most important in transplants, platelet transfusions, and some transfusion reactions (TRALI, TAGHVD and some febrile rxns)
-- Bennett-Goodspeed (BG) ags found in small amts on RBC surface
- 50% chance that 2 offspring from same parents will be haploidentical (basically a Punnett square)
MHC divided into 3 classes:
MHC class I found on all nucleated cells and platelets, but only very little seen on mature RBCs
- Class I genes found on loci HLA-A, -B, and -C, which can further be broken down into HLA-A1, -A2 etc
-- each gene makes a protein similar to an Ig heavy chain that gets embedded through a cell's membrane and is assoc c B2 microglobulin
- put "w" superscript after HLA-Cw to distinguish from complement
- a star ( * ) after HLA means that it was determined by DNA (not serologic) methods
MHC class II found on B cells, activated T cells and macrophages, activate Class-II specific CD4+ T cells
- encoded by genes at loci called HLA-DR, -DP, and -DQ, each of which can have multiple alleles (HLA-DR2, HLA-Dw3); genes make an a and B protein similar to Ig light chain that embeds in cell wall
MHC class III not important in blood banking (codes for complement proteins)
- genes for hemochromatosis, 21-hydroxylase and TNF also found in the MHC gene region
"Haploidentical" means that 2 people share one HLA haplotype (one cr 6 is the same)
Linkage disequilibrium - when the expected and observed frequencies of alleles do not match

on the surface of grp O







Acquired B phenotype; BBGuy















Antibody Identification
ID done after pos ab screen on pts, prenatals, donors
- can be if tests show new ab or confirm prev id'd ab
Alloantibody - ab against RBC ag not present on pts own RBCs
Autoantibody - ab against RBC ag present on pts own RBCs
Panels are expanded ab screens
- use grp O reagent RBCs (8-20 donors), pt serum / plasma
- tubes use IS / 37C / AHG, gel/solid phase only AHG
- rxns documented on sheet



e
cell 1
cell 5
cell 2
null Duffy A


PEG not read at 37C bc of nonspecific rxns


should be 0, otherwise it is reacting against itself
General checklist
1. Check hx (up to 7/10 cases impacted by HX!!)
ex:
- anti-D in preg pt; consider RHIG
- recent bacterial infx; consider warm antibiotic-induced warm autoab
- recent viral illness: consider auto- anti-I or -i (look at the age)
- recent transfusion: consider newly developing ab
- ITP: consider IV RhiG in D+ pt
- consider racial profiling: blacks lack Duffy; almost all Asians are D+; whites can lack high freq ags
- check for serologic hx
- previous phenotyping helpful, but be careful of transplants, transfusions, errors (can help target an ab)
2. Check autocontrol (AC)
- consider if DAT - pos and look at the pt Hx;
- may be 2/2 autoabs (warm and cold), recent transfusion / DHTR, drug-induced, passively acquired abs
3. Look at general pattern
Uniform or variable?
- uniform rxn suggests single ab; variable rxns can mean either multiple abs OR single ab c dosage
Against all, most or rare cells?
- with neg AC, mix of reactive and nonreactive cells suggests a single alloab OR multiple alloabs;
- single reactive patterns suggest single alloab against a low-prevalence ag
- all cells reactive means multiple alloabs or a single alloab against a high-prevalence ag
Present in what phases?
If present at IS and 37C, consider a newly developing ab that is still in its IgM phase
- cold phase is actually room temp.t
4. See what is NOT there (cross-outs)
5. Look at what IS there
- try first for a single ab to explain all rxns
6. Use special techniques as necessary
7. Ensure statistical significance







First, looking at the cells that didn't have a rxn, go down the line starting at the rt, ruling out abs c pos rxn








6. Use special techniques as necessary
- helps confirm id of alloab by demonstrating lack of ag
- is a TOOL in confirmation, not sole measure of confirmation
Adsorption - removing abs from sample by incubation c ag-pos RBCs
Elution - removal of RBC-bound abs; heat, cold, chemicals (glycine)
Proteolytic enzymes - as in ficin and papain that can change ag expression / binding
7. Ensure statistical significance
- must ensure that what is observed is not 2/2 chance
- Traditional enterpretation:
3 pos rxns = ag present
3 neg rxns = ag absent
- for practical purposes, prelim ID only requires 1 double-dose rule out to establish lack of id
-- must still use selected cells to rule in or out specific abs (as in ur OP)

BB guy notes


Crossmatch
Can be done either serologically or by computer, basically just a final check for ABO compatibility or to see if significant alloantibodies present
- when done serologically, sample of recipient serum is mixed with donor blood (from the heat clamped tubing) and spun at room temp, and is thus called an Intermediate Spin (IS)
-- may be warmed to prevent "nuisance" cold antibodies
-- if clinically significant alloantibodies present then must add a second antiglobin step
- if done by computer, it must be validated and ABO must be done twice
Red Cell Panel
Basically an in-depth ab screen, used when ab screen or crossmatch is (+), uses 10 different types of cells
- used to see which abs are causing the rxn (and if significant) and what would be the best transfusion donor; done in agglutination testing to find RBC clumping from ab coating that causes coating of cells (sensitization) and formation of bridges if positive
- tests carried out in enhancement media such as Low Ionic Strength Saline (LISS; decreases repulsive charge bwt RBCs; enhances cold abs) or polyethylene glycol (PEG; excludes H2O, enchances warm abs)
- positive result is either hemolysis or agglutination; though must check to see if controls react or not
The first stage of agglutination is coating of cells (sensitization), which depends on ab specificity, RBC electrostatic charges, temp, amts of ag and ab
- the second stage of agglutination is the formation of bridges (a lattice structure c abs and RBCs), which IgM is best at, IgG not as much
- hemolysis also read as a positive result, but is less common (requires complement fixation, IgM still better)
Tube testing
3 phases:
(1) Immediate Spin (IS) phase done by centrifuging a mix of 2 drops of serum and 1 of RBCs at room temp for 15-30 seconds; reactions usually 2/2 IgM (not usually significant)
(2) 37 C phase tested c anti-IgG reagent or polyspecific reagent in AHG phase (potentiators like LISS or PEG) and then incubated for 10 min to 1 hour (dpending on the potentiator) and then spun
- not very useful by itself (sometimes not done; and in general should not do a PEG read at 37C)
(3) Antihuman globulin (AHG) / Indirect antiglobin test (IAT) - Wash same tube used for 37 C, add AHG and centrifuge (washing remove unbound globulins that "neutralize" (bound by) AHG that cause false-neg rxn)
- only req'd part of ab detection bc best for detecting warm IgG abs; polyspecific or IgG- specific AHG can be used (lab preference)
Grading done on scale of 0-4+; 4+ is a tight cell button and negative (0) is smooth, easily dispersed RBCs
Column (gel) agglutination
Microtubes are coated c IgG which can stop the migration of RBCs through a column which has similar sensitivity to PEG tube testing
- mix RBCs and plasma at top of chamber above column filled c gel and anti-IgG
- essentially skips the 37C incubation phase
- gel agglutinates based on size and binding to IgG-coated RBCs
-- strong positive agglutinates at top of gel
-- complete negative gives RBCs at bottom of gel
- similar to PEG-enhanced tube testing, is excellent at detecting warm auto-abs (and has similar sensitivity)
Solid-phase red cell adherence testing, where abs bind to lysed or intact RBC ags that bind to the sides of microwells, and then pt serum added and read as positive result if form a carpet along the well and negative if forms a button on the bottom
- negative is the solid button at bottom of well (meaning there were no attached plasma antibodies that anti-IgG-coated indicators cells could bind)
- strong positive (4+) has diffuse carpet of indicator RBCs spread all across bottom of well, indicating that the plasma ab attached to the well-bound RBC ags
Cold antibodies are IgM and are insignificant (except for ABO group incompatibility) whereas warm abs are IgG and are significant
*** Guadalajara is warm; Michigan is cold***
- significant abs can cause HDN or hemolysis
-- anti-M, N, Lewis, Lutheran and I are nearly always insignificant
Antiglobulin Testing ("Coombs test")
Direct Antiglobulin Test (DAT) - AntiHuman Globulin (AHG) is added to washed blood from pt; (+) if agglutinates; is basically the last step of the IAT
- shows in vivo RBC coating c ab or complement
- positive DATs are non-specific and seen in up to 15% of hospitalized pts
- may be useful in transfusion rxns, auto-abs and AIHA, HDFN, drug-assoc hemolytic anemia, abs vs recently transfused ags
- Ab binds RBCs and causes agglutination w/o further manipulation
- IgM best at this bc diameter wide enough to overcome zeta potential, whereas IgG can do this if ag sticks out far enough from RBC surface (ABO, M and N ags most commonly)
Indirect Antiglobulin Test (IAT) - pt serum (with abs) added to uncoated RBCs, along c AHG
- checks in vitro RBC coating
- Unknown Ab Checks (use RBCs c known ag profile; as in ab screen) or Unknown RBC Ag checks (use serum c known ab spec; as in RBC ag testing) are IAT variations that can be used to check unknown abs or ags, such as in crossmatch procedures
- Abs bind but do not form bridges c RBCs and needs another step to agglutinate (enzyme tx can make IgG capable of direct agglutination
- classically IgG not IgM (most significant abs cause this type of agglutination)
In pretransfusion testing, pt serum added to solution of donor RBCs to check for incompatibility bwt recipient abs and donor RBCs
- can be done c known serum ab and unknown RBCs or with RBCs of certain phenotype to check for serum ab
Types of anti-human globulin (AHG)
(1) Polyspecific (polyclonal anti-IgG and monoclonal anti-C3d)
- previously the MC AHG, but less popular now
- if +, labs then test c anti-IgG and anti-C3d separately
(2) anti-IgG
- used for gel and solid-phase platforms exclusively
- lots of labs use anti-IgG in tube tests only
- can have some cross-reactivity c other Ig types 2/2 rxn c kappa and lambda light chains shared by Ig's
(3) anti-C3d
- C3d is a nonreactive byproduct of complement fixation on RBCs
- anti-C3d useful to eval IgM-related hemolysis and cold agglutinin dz, where abs not usually detectable via anti-IgG
Specificity possibilities for the antiglobulin
1) Anti-IgG, -C3d (“polyspecific”); most common to start
- Detect red cells coated with either of the above
- May also detect other immunoglobulins (because
the anti-IgG detects light chains, too)
2) Anti-IgG and anti-IgG (heavy chain-specific)
- Both detect IgG-coated red cells
- Anti-IgG used for PEG, gel, and solid phase tests
3) Anti-C3b, -C3d
- Detects either of above complement components
- Most useful in evaluating IgM-related hemolysis,
cold agglutinin disease
IgG-sensitized RBCs (“Coomb’s control”, “check cells”)
a. Use after negative DAT or IAT tube test (not gel or
solid-phase) to ensure functioning of AHG reagent
b. Add IgG-coated cells to AHG-cell mixture, and should see free AHG agglutinating the check cells
c. if Negative = bad AHG or no AHG added
d. Other errors (e.g., omitting test serum) missed.
- solid-phase tests run a positive control in parallel, so no additional AHG control is req'd
Abs display dosage
if they have variable strength of reactivity or they react across the panel board
- usual suspects are MNS, Kidd, C/c, E/e, and Duffy
- Some abs react more strongly with RBC antigens
that have homozygous gene expression.
-- For example, imagine a hypothetical anti-Z
a. Patient 1 genotype: ZZ (Homozygous for Z)
b. Patient 2 genotype: ZY (Heterozygous for Z)
- if anti-Z shows dosage, it will react stronger c pt 1's RBCs (such as a 3+ rxn in Z homozygous, 1+ ZY)
High Titer / Low Avidity (HTLA) abs usually react weakly against all cells in a dilution, and at titers >1:64
- significant: Cartwright (Yt^a), Holley (Hy), Gregory (Gy)
- not significant: Chido/Rogers (Ch/Rg), Sda, Bg, Csa, York (Yka)
T-activation - bacterial neuraminidase activates T, Tn and Tk ags on RBCs, for which people usually make IgM abs
Elutions can be taken from pts serum to test what ab it contains
- hemagglutination inhibition is using a fluid known to contain a mimic of a certain ag, and is used to determine if it neutralizes an elution with a suspected ab
Enzymes (such as papain, trypsin, ficin) cleave RBC surface glycoproteins and can strengthen rxns by allowing abs to bind better to previously shielded ags and can thus be added to see if antigen are enhanced or destroyed
-- Le, I/i, P, Rh and Kidd typically enhanced
-- MNS, Fy^a/b, Lutheran, Chido, Rodgers and Yt^a usually destroyed by enzymes
- Kell, Diego and Colton are unaffected
- similarly, certain substances can neutralize abs (see table from BBGuy)
Lectins are seed / plant extracts that react c certain RBC ags and can be esp useful in polyagglutination (T, Tn, etc); can be commercial or homemade
Prewarming
Pretransfusion testing c all reagents and samples incubated and kept at 37C can help eliminate effects of cold auto- or alloabs
- is NOT a way to get rid of reactivity of stuff you don't understand!!! Can weaken significant abs and should only be used as confirmation of workup already performed
Adsorption
Removal of specific abs from sample via incubation c ag positive RBCs
- used to remove warm or cold auto-abs ("autoadsorption") from sample to detected allo-abs
- can also remove 1 or more allo-abs ("alloadsorption") from sample to detect or confirm other alloabs
- can be used c multiple abs to clear a muddy picture
- ie. sample has anti-K, anti-C, and anti-S, but anti-S not well visualized; can use K+C+S- RBCs to adsorb the anti-K and anti-C and leave the anti-S in the "adsorbed serum" for clearer results
Elution
technique for removal of abs bound to RBC surface for analysis
- can be done c heat, cold, chemical (ie glycine) tx
Dithiothreitol (DTT) or 2-mercaptoethanol (2-ME)
- denatures surface RBC ags of multiple groups (Kell, Lutheran, Dombroc, Yt, LW)
- can also remove IgM ab activity from serum
ZZAP
Combo of DTT and proteolytic enzyme
- acts on combo of enzyme sensitive and DTT-sensitive ags
Chloroquine
Removes IgG from coated (DAT-pos) RBCs to allow for accurate phenotyping (effective at least 80% of the time)
- also removes residual HLA ags from RBCs (Bg ags)
Autoantibodies
Think of them c (+) DAT or (+) control
- (+) DAT observed in up to 3/20 hospitalized pts
- may not be significant if pt not hemolyzing
- rx, recent transfusion and other causes may also cause a (+) DAT
-- rx can provoke DAT (+) by: haptens (rarely seen c penicillin); non-immune protein adsorption (cephalothin); immune complexes (quinidine, cephalosporins - RBCs test + for C3d); or true AIHA (procainamide and aldomet)
Warm Autoantibodies
Usually IgG, react at 37 C, cause (+) DAT, hemolysis called Warm AutoImmune Hemolytic Anemia (WAIHA) [most warm abs do not cause hemolysis]
- strength of positivity in AHG phase can correlate c chance pt will hemolyze
- MCC is ab c broad anti-Rh specificity
- best to avoid transfusion and try to find out the ab
-- if necessary, can do autoadsorption procedure to remove abs from serum using ZZAP, then do screen on that adsorbed serum
Cold Autoantibodies
MCC autoantibodies in general and MC is anti-I, least common is anti-i; have wide temp range; mostly IgM
- Cold AutoImmune Hemolytic Anemia (CAIHA) MC in old people c Raynaud's phenomenon and chronic hemolysis
- can occur for 2-3 weeks after M pneumonia infx
-- mono can cause IgM anti-i
Paroxysmal Cold Hemoglobinuria (PCH) seen in kiddos c viral illness or syphilis after cold weather exposure who present c hemoglobinuria, fever, back pain, jaundice, severe anemia
- tx: keep that kid warm
- lab must use Donath-Landsteiner test to find IgG biphasic (warm and cold) hemolysin c anti-P specificity
Mixed-type autoabs, with both IgG and IgM can occur in lupus pts, reacting c both IgG and C3 in AHG phase, and can be tx'd c roids
Neonatal / Intrauterine Transfusion
Intrauterine transfusion requires fresh, washed, irradiated, type O and Rh negative (and other ag neg) blood
Neonates need compatible products for FFP, platelets or anything with a substantial amt of plasma
- vol reduction not necessary in neonates bc it may complicate things (infx, delay in transfusion, plt loss)
Maternal Immune Thrombocytopenic Purpura (ITP)
Auto-abs can cross placenta in mamas who have had ITP in the past, causing TBCpenia in baby
- should monitor plts in baby's at risk, can (rarely) get TBCpenia or intracranial bleeding
- may give plts or IVIG in babys c sx, though plts should improve as they stop getting abs from mama
Neonatal Alloimmune Thrombocytopenia Purpura (NATP)
Similar to above, caused by maternal abs crossing placenta causing TBCpenia (<20k) and causing a higher incidence of intracranial bleeds and in utero bleeds; can happen in first pregnancy
- tx c early cesarian delivery and plt transfusion from mom; IVIG and roids usually don't help too much
Hemolytic Disease of the Newborn (HDN)
Hemolysis caused by IgG (1, 3, or 4)allo-abs crossing placenta and leading to anemia, hyperbilirubinemia, hydrops fetalis
- now MC caused by abs other than anti-D to Rh (MC is ABO, but anti-Kell > anti-c are the most severe)
- anti-D titer needs to be >1:16 to be significant
RhIG can be given to mom at risk of developing anti-D abs, usually at a given time interval (@28 weeks and post-partum [at least 1 vial]) or after fetal-maternal hemorrhage
- Kleihauer-Betke test or ELAT used to determine the amt of fetal blood in mamas circulation, and thus the number of RhIG vials that needs to be given
Various forms (IM or IV) or RhIG
RhoGAM, Rhophylac, HyperRHO, WinRho
US full dose vial (300 um/ 1500 IU) : 30 mL D+ blood or 15 mL RBCs)
- mini-dose vial (50 ug/ 200 IU) for 5mL D+ blood or 2.5 mL RBCs)
If Fetal Bleed Screen neg, give 1 vial
- count the number of rosettes, if >3 / 10 hpfs
- in KB test, citric acid added and mom's cells fade, bc HbF from baby is resistant to acid, while HbA elutes
- if decimal is 0.0-0.4 round up 1 vial
- if decimal is 0.5-0.9, round up 2 vials
Sickle Cell Disease Transfusion
Exchange transfusion in sickle cell pts should be considered to reduce risk of iron overload
- alloimmunization also common 2/2 inc amts of transfusions
- transfusion goal in these pts based on HbS levels (<30% kiddos, <50% adults), not on Hgb levels
- 1/10 pts get a stroke, can reduce risk c chronic transfusions; risk of 2nd stroke greatly increases after 1st stroke
- ~1/2 of chronically transfused pts get alloimmunized at rate of 3% per transfusion, usually to Kell, C, E, and Jkb (can be greatly reduced if products selected for these abs)
Massive transfusion
Massive transfusion = replacing total blood vol in 1 day
- transfused blood does not immediately carry oxygen well 2/2 left-shifting of the oxygen dissociation curve from 2,3- DPG depletion; also lowers blood pH, inc K+, and can cause dilutional coagulopathy
If you have 30 min, should try to crossmatch blood products, otherwise give O-neg



Solid-phase testing


Can Med Assoc Journal Jan 2006

Reading tube-testing








*** My Dog Lassie**
*** A Rotten Kid ***


HDFN





+ rosettes on fetal blood screen

Fetal Blood Screen
Blood Products
Whole blood
Not widely available; 500 cc of RBCs (Hct 40%), plasma, WBC, and platelets
- stored at 1-6 C, time stored varies c preservation
- may be indicated for exchange transfusions (neonates)
- contraindicated if pt at risk of vol overload
- pt outcome worsens c each unit of RBCs transfused bc of infx rates, inc length of stay, morbid and mortality
Packed Red Blood Cells (pRBCs)
300 mL of RBCs (Hct 55-80%), minimal plasma, WBCs and platelets (250 mL total) and 200 mg iron
- given for decreased O2-carrying capacity; increases the Hct by 3% (can only infuse in same line with saline, no meds or other products)
- also stored at 1-6 C, storage time varies c preservation (keep at 1-10 C during transportation)
-- able to be given as long as 3/4 of the RBCs are viable, the bag is not spiked, the temp has not risen substantially, and there is >= 1 sealed segments left
-- preservatives contain citrate (in CPD/A) to soak up Ca2+; a sugar (dextrose in all) or adenine (in CPDA) for ATP and sodium phosphate to control pH
- should not give if pt is hemolyzing (?)
- factors V and VIII decrease over time
- dec 2,3-BPG and pH causes right shift in O2 dissociation curve, enhancing O2 release from RBCs
Max storage time depends on preservative:
CPD - 3 weeks
CPDA - 5 weeks
AS - 6 weeks (42 days)
-- AS-1 (Adsol), AS-3 (Nutricel); AS-5 (Optisol)
-- “AS” = Adenine-saline
-- Result in a product with more adenine for ATP generation
-- AS-1 and AS-5 have mannitol for red cell preservation (not for diuresis)
- AS-7 (aka SOLX) has bicarbonate
- RBCs collected in CPD can be rejuvenated up to 3 days after expiration and frozen up to 10 years (RBCs must be washed before being used after rejuvenation to remove the rejuvenation soln)
- older units have less viable # RBCs (~80%), lower pH, dec ATP, 23-DPG, and inc plasma K+ and Hgb
Irradiation prevents transfusion-assoc graft-versus-host disease (TA-GvHD), which wipes out the bone marrow and kills in about a month (causes a rash, T cells invades liver, GI, and bone marrow [kills immune system])
- crosslinks DNA of WBCs (esp T-lymphocytes) and makes them not capable of proliferation
- all HLA matched products MUST be irradiated bc viable donor lymphocytes may be homozygous for HLA haplotype for which recipient is heterozygous
- irradiated units expire 28 days after irradiation or at their original expiration date (whichever first)
- needs minimum of 15 Gy at periphery of radiation field and 25 Gy at center, with the time depending on the half-life of the source (which is shielded, thus no extra precaution needed for pregnant technologists)
Indications: T-cell defects (aqd/congenital), HSCT, intrauterine exchange transfusions, premature infants / low birth weight (<1200 g) newborns, Hodgkins, transfusion from blood relatives, transfusions from HLA-matched
- in granulocyte transfusion, fludarabine / purine analogs
- probably should do c neonatal exchange transfuisions (esp if IUT), other leukemias, possibly solid tumors (neuroblastoma), cardiac surgery
DO NOT need: solid organ transplants (even if on immunosuppression), small vol tx to neonates (-IUT), HIV/AIDS pts, recipients of prev frozen products (such as FFP since freezing inactivates lymphs), though is needed in fresh plasma
Leukocyte reduced is NOT the same as irradiation
- do NOT do on stem cell transplants
Can store for 28 days after irradiation
- irradiation does not make product radioactive!
Leukoreduction can be done to prevent febrile rxns (1-log reduction), HLA immunization / CMV infx (3-log reducction [WBCs with the virus are removed])
- deglycerolized RBCs (glycerol added when freezing RBCs at -80 C [lasts up to 10 years] to prevent damage) is same as leukoreduced
- Europe leukoreduces all units (definition of leukoreduced is <1 mil WBCs in a unit); >99% of units in the US meet this European standard, but we just don't mention it
- LR utility is limited, may not be cost-effective (it costs a lot)
- filter failures can occur (esp in pts c sickle cell)
- leukoreduction does not protect against TA-GvHD
WBCs degenerate and undergo necrosis and apoptosis and secrete substances into the plasma that can cause febrile transfusion rxns
- can also present foreign HLA ag to the host immune system, which leads to anti-HLA abs, causing plt refractoriness, issues c transplants and fever
- Transfusion-related immune modulation (TRIM)
Washing RBCs (resuspension in saline) removes plasma to prevent allergic rxns
Autologous donations should be done 3 days before a procedure
The activity based cost for providing blood is about 3-4x higher than the direct cost of acquiring the product ($200 acquisition cost), bc of product testing, reagent costs, lab and nurse time to prepare and admin product and bc of cost of supplies
Platelets (random donor)
50 mL of > 50 bil platelets, a little bit of plasma, WBC and RBC
- stored at 22-24 C, lasts for 5 days
- might not want to give in ITP, do not give in TTP
Platelets (pheresis)
300 mL of > 300 bil platelets, and a little bit of plasma, RBC and WBC
- stored at 22-24 C, lasts for 5 days
-- can last for 21 days with Acid Citrate Dextrose (ACD)
- might not want to give in ITP, do not give in TTP
- diversion of the first few milliliters of blood in pouch is proven method to dec risk of bacterial contamination by capturing the "skin plug" and any bacteria in the plug
- 90% of the units should have >3 x 10^11 platelets (and < 5 x 10^6 lymphocytes in 95% if leukoreduced)
Fresh Frozen Plasma (FFP)
200 cc of plasma (containing all coag factors), 400 mg fibrinogen
- stored at -18 C (very cold!!), but lasts for a year
-- to use, thaw and centrifuge @ 1-6C and freeze the precipitate @ -18C within an hour of preparation
- no contraindications
- plasma frozen in the first 24 hours, but after the 8 hour limit that must be frozen in to call it FFP is a specific plasma product called PF-24, but is thought of virtually the same as FFP
- plasma that has had cryoprecipitate taken from it (called Cryo-Poor Plasma or Cryoprecipitate Reduced Plasma) can be used in the tx of TTP, since it would be devoid of vWF, but is as good as FFP in Therapeutic Apheresis
- indications for transfusion of FFP are weak, but may be given for massive transfusion or reversal of warfarin anticoagulation in pts c intracranial bleeding
- may help to correct INR, PT, PTT immediately before surgery
Cryoprecipitate
15 cc of the cold-insoluble part of plasma that has at least 150 mg of fibrinogen, 80 IU of factor VIII and a little bit of factor XIII and vWF and fibronectin
- must have >80 IU factor VIII and 150 mg fibrinogen
- like FFP, stored at -18 C, lasts a year, and has no contraindications
-- FFP is thawed and centrifuge @ 1-6C and the precipitate frozen @ -18C within an hour of preparation after resuspension in saline
- may be necessary to give cryoprecipitate after plasma exchange, which can remove ~60% of the bodies fibrinogen and overload the liver's ability to make it
- may be given for fibrinogen def, or tx of von Willebrand dz
Thawed cryoprecipitated AHF expires in 4 hours in an open system or pooled, and 6 hours if thawed and not pooled
Granulocyte concentrate (pheresis)
200 cc pus bag (has over 10 bil granulocytes)
- stored at 22-24 C but only lasts a day
- no contraindications
- irradiation can be done on a unit for an immunocompromised pt to prevent GVHD
Stem cell transplantation
Bone marrow transplant (BMT) and peripheral blood stem cell transplant (PBSCT) have similar overall survival and disease free survival
- BMT has longer time to neutrophil and platelet engraftment, but has lower rates of GvHD
- PBSCT can have side-effects from agents used to mobilize stem cells
Extracorporeal photopheresis
Cutaneous T-cell lymphoma is the only FDA approved indication for extracorporeal photopheresis
Transfusion assoc Infx
Three main barries from the donor, to transmitting a dz to pt
1) Education / self-deffereal
2) Screening questions
3) Donor testing
per FDA: Donor screening donor deferral lists, donor blood testing, quarantine until OK, quality practices (GMP)
A big wall bwt donor and pt is Pathologen reduction and inactivation
- direct tx of blood products,
- hear, solvents - detergents, methylene blue
- direct nucleic acid maipulation (already in EU, not USA)
-- intersect system deactivates viruses and bacteria readily; known and unknown
-- probably same for parasites
- effective WBC deactivation
-- dec need to irradiate, leukoreduce, or culture
Test methods
- Enzyme Immunoassay (EIA)
- Chemiluminescent immunoassay (ChLIA)
- Western blot (WB)
- Immunofluorescnece assay (IFA)
- Recombinant immunoblot assay (IBA)
- Nucleic acid testing (NAT)
Enzyme Immunoassay (EIA)
- Enzyme-linked immunoassay (aka ELISA or Immunoassorbant assay)
- developed in 70's, majority of ag testing
- some ag testing (HBsAg)
- very sens and spec, tho majority of (+) tests are false-pos
ChLIA
Very similar EIA, except the detection method (acrodynium) emits light
Western Blot (WB)
HIV confirmation after reactive EIA
- similar to EIA in principle
- proteins separated onto membrane by electrophoresis
- ab (if present) binds
- anti-human Ab binds and is detected
- Problem: indeterminates
IFA
Good alternatiave for indeterminate WB samples
RIBA
- HCV confirmation after reactive EIA
- similar to WB
- recombinant Ag on strip (MFG)
- same ags as current EIA req'd
- Abs bind
- peroxudase-labeled anti-human Ab
- H2O2 added
NAT
- dont say NAT testing!
-PCR and TMA two main vars
- uses molecular techniques to find, amplify and detect specific genetic material
- very sens and spec
- invented in 1983 on PCH
PCR
amplified selected genetic area
- seq: denaturing
TMA
- faster, may be more sens than PCR
- theoretically simpler, less contamination, no thermocycling
- RNA or DNA target
- RNA amplicon
Terms
- Window period - infx to lab detection
- Eclipse phase - entry into cell to new virus appearance
- incubation period - exposure to clinical sx
-- window period and incubation period usually pretty similar
Reactive?
- initially reactive - first screening test (generally EIA / ChLIA) value exceeds cutoff; the next step is repeat in duplicate
- if repeat reactive, one or both repeat tests also reactive; unit always discarded
Standard 5.11.3.6 = cellular therapy products must be tested for CMV
- a pos test, however, does not preclude donor from donation to CMV pos or neg recipient

Transfusion Reactions
Opinion 1: Assume all suspected reactions are hemolytic, and work to disprove your assumption:
1) A high index of suspicion is much better than low
2) You will be wrong in your assumption almost every time, but the one time that you are right will be life-saving!
Opinion 2: Anyone involved in a transfusion should be allowed to initiate a transfusion reaction workup
1) Nurses, perfusionists, and other transfusing staff should be empowered to contact the blood bank directly if suspicious findings occur during transfusion
2) This is from hard experience, with suspected reactions being under-reported (including the worst acute HTR of my professional life)
STEP ONE: STOP THE TRANSFUSION!
a. Don’t disconnect the unit (though that will eventually happen); at least stop the incoming flow of blood.
b. Main indicator of survival of an acute HTR: amount of incompatible blood infused; as a result, the obvious thing to do if you are assuming hemolysis is to stop the transfusion
c. Leave the line open with saline.
Necessary checks
a. Clerical check
1) Bedside paperwork and bag check to ensure right unit went to right patient
2) Blood bank paperwork and computer check to answer same question
3) Should include a basic inspection of the unit for discoloration or obvious issues
a) Darkened color in unit: Suspicious for bacterial contamination
b) Check for clots, aggregates, or anything out of the ordinary
b. Visible hemoglobinemia check
1) Spin a post-transfusion EDTA sample and examine visually for a pink-red color change indicative of free hemoglobin in the plasma
a) Best to use EDTA because you can then use the same sample for the tube DAT
2) Compare to pretransfusion sample if abnormal.
3) Detects as little as 2.5 to 5 ml of hemolysis occurring anywhere in the body
4) Most sensitive way to detect intravascular hemolysis; not specific, though
a) Some causes of false-positive visible plasma hemoglobin:
• Poor phlebotomy technique (traumatic stick, drawing through IV line)
• Nonimmune hemolysis (infusion with 0.45 NS, faulty blood warmers, etc.)
• Autoimmune hemolysis
• G6PD deficiency and hemoglobinopathies
b) Some causes of false-negative visible plasma hemoglobin:
Delay in drawing sample (with functioning kidneys, hemoglobin may be cleared in several hours)
Sample collected from IV line (dilution of blood)
c. Direct antiglobulin (Coombs) test (DAT)
1) Demonstrates coating of RBCs with antibody and/or complement in-vivo (see figure 3 below)
2) Most commonly done with polyspecific method (IgG + C3d)
3) If positive, must compare to pretransfusion DAT (automatically done in most places)
4) Note that a positive DAT does not prove an acute hemolytic reaction
a) Other causes include nonspecific positives in hospitalized patients (20%), autoantibodies, drugs, passive administration of other things like RhIG or IVIG
5) Also note that a negative DAT does not disprove an acute hemolytic reaction
a) If donor RBCs are completely destroyed by brisk hemolysis, DAT will be negative (especially seen with ABO incompatible transfused RBCs)
b) Small amounts of residual donor RBCs (<10% of circulating RBCs) may give false negative tube DAT; gel DAT may help if strongly suspected
d. Repeat ABO/Rh testing
1) Another check for right patient, right blood
2) Check both pre- and post-reaction specimens
5. Other things that may be done (but are not required as a part of every workup)
a. Facilities must define triggers for when they would do additional testing
Additional testing for suspected septic reactions:
1) Not done routinely on all reaction workups by most facilities
2) Should be done if suggested by clinical data, for example:
a) Temperature greater than 102 F
b) Temperature increase greater than a specified amount (2 or 3 degrees F)
c) Severe rigors
d) Clinical septic shock-type findings (marked hypotension, gastrointestinal complaints, later findings of multi-organ failure, DIC, etc.)
3) Both patient and product must be evaluated
a) Patient:
• Blood cultures (drawn as soon as possible from a different site than the infusion); aerobic and anaerobic
• Consider culture of all intravenous fluids running at the time of reaction if clinically suspicious of sepsis
b) Product:
• Gram stain and culture of actual residual product in the bag (don’t culture a segment unless there are absolutely no other options!)
d. Additional testing for suspected respiratory reactions (see details in TRALI/TACO sections):
1) Chest X-ray
2) BNP levels
3) ABG
4) Donor testing for anti-HLA/HNA antibodies
e. Additional testing for suspected severe allergic reactions:
1) Serum IgA levels (performed on pretransfusion sample!)
2) Consider serum anti-IgA detection if serum IgA is non-detectable
C. Classification of reactions
1. Worldwide, there is a movement to standardize the definitions of transfusion reactions as well as to improve reporting
2. In the US, this movement is led by the CDC National Healthcare Safety Network (NHSN)
and is known as “Biovigilance” (more specifically as “hemovigilance”)
3. This lecture includes entities defined in the hemovigilance module (found at
http://www.cdc.gov/nhsn/TOC_BIOManual.html)
4. Below is an approach to screening transfusion reactions based on the presence or absence
of fever and the timing of the reaction (Acute = during or < 24 hrs after transfusion, Delayed = > 24 hrs after transfusion)
Acute reactions presenting with fever
Acute hemolytic transfusion reactions(AHTRs)
Incidence: 1:76,000 transfusions, (1:1.8 million transfusions fatal HTR)
Clerical errors (both in transfusion service and at bedside) are most common cause
RBC destruction may be intravascular or extravascular
1) ABO-related, intravascular usually more severe
2) 2009-11: More US non-ABO-related than ABO-related fatal HTRs
Signs/symptoms
1) Timing
a) Severe reactions may occur early in transfusion (first 15 minutes; see figure 4) b) Milder reactions may present later, but usually before end of transfusion
2) Specific signs/symptoms:
a) Fever and chills
• Most common presenting symptom (> 80%)
b) Back or infusion site pain
c) Hypotension/shock
d) Hemoglobinuria (may be first indication of hemolysis in anesthetized patients)
e) DIC/increased bleeding (also important in anesthetized patients)
f) Sense of “impending doom”
Lab findings
1) Hemoglobinemia (pink or red serum/plasma); lasts several hours in those with adequate renal function
2) Hemoglobinuria (typically clears by the end of one day)
3) Positive DAT (unless all donor cells destroyed); may be “mixed field”
4) Elevated indirect and direct bilirubin
5) Lab findings of DIC (D-dimers, decreased fibrinogen, etc.)
6) RBC abnormalities
a) Schistocytes: Intravascular hemolysis
b) Spherocytes: Extravascular hemolysis
f. Pathophysiology
1) Intravascular hemolysis due to ABO incompatibility typifies these reactions
a) ABO antibodies fix complement well (high antigen density, potent IgM/IgG antibodies), and this leads to membrane attack complexes and rapid RBC lysis
b) Other antibodies (especially Kidd) may also fix complement and lyse RBCs
c) Seen much less commonly with incompatible donor plasma (e.g., platelet transfusions from group O donor with high-titer anti-A to group A recipient)
2) Hemolysis leads to a complex array of events, including:
a) Release of free HGB and HGB-free RBC stroma into circulation
b) Stimulation of intrinsic coag pathway and bradykinin via Ag-Ab complexes
c) C3a and C5a generation (“anaphylatoxins”)
d) Production of several very important cytokines:
• Tumor necrosis factor (TNF-α)
• Interleukin-1β (IL-1β)
• Interleukin-6 (IL-6)
• Interleukin-8 (IL-8, aka CXLC8)
4) Extravascular hemolysis (e.g., Rh/Kell/Duffy, etc.) is usually but not always less severe due to lack of systemic complement and cytokine activation
Treatment
1) Hydration and diuresis are critical early components for hypotension treatment and renal function preservation
a) Maintain urine output > 1 mL/Kg/hr with saline +/- furosemide
b) Low-dose dopamine use is controversial (may not preserve function)
2) Consider DIC; some use heparin during hypercoagulable phase of DIC
3) Consider early exchange transfusion, esp. for high-volume incompatible transfusion
Prevention possibilities
1) Training and careful attention to phlebotomy, labeling, issue, and administration
2) Some require two separate ABO/Rh types before transfusion
3) Advanced methods (RFID, bar codes, etc) will likely be helpful in future
Febrile nonhemolytic transfusion reactions (FNHTRs)
Historically most frequently reported reaction
1) Incidence decreased since “universal” leukoreduction (0.1 to <1%)
2) Still more common than any reaction in heme-onc and transplant patients, especially with platelet transfusion (before LR, as many as 1/3 in some studies!)
Unexplained increase in temperature of 1C or 2oF
1) Not intended to be a strict cut-off; i.e., don’t require it to diagnose or work up rxn
Cause: Increased pyrogenic substances (e.g., TNF-α, IL-1β, IL-6), mostly from WBCs
1) Cytokines may be produced before transfusion
• Donor WBCs secrete while in the storage bag.
• Transfusion simply infuses pre-formed fever-inducing substances
• More common in platelet transfusions
2) OR, cytokines may be produced after transfusion
• Recipient anti-HLA/HNA antibodies attack donor WBCs, or (less commonly)
donor antibodies attack recipient WBCs
• This action leads to fever-inducing substance release after transfusion
• More common with RBC transfusions
Signs/symptoms
1) Transient fever and chills (+/- rigors?) during or up to 2 hours after transfusion
2) Symptoms tend to occur later in transfusion; if very early, be suspicious of transfusion-related sepsis
3) Note that chills may be first; fever may be delayed up to one hour or more after transfusion in up to 10% of cases
4) Variant versions in premedicated or head injury patients may never have fever
Differential diagnosis:
1) Acute HTR
a) Remember the above caveat that most AHTRs present with fever/chills alone
b) Impossible to distinguish early AHTR from FNHTR clinically, though most fevers in transfusion will NOT be AHTR
2) Transfusion-related sepsis
a) Especially sepsis from PLT transfusions (RBC-related sepsis usually presents earlier in transfusion)
b) Timing may be identical, mild temperature elevations may mean underdiagnosis of PLT-related septic reactions
Transfusion-related sepsis (septic transfusion reaction, bacterial contamination)
General statements
1) Bacterial contamination is the #1 infectious risk from transfusion, much more common than viruses
2) Some sources: As many as 1 in 3000 platelet units are contaminated (many fewer reactions, however)
3) Most contaminated products that cause reactions are closer to their expiration date than their collection date (gives bacteria time to proliferate and enter log phase)
b. How does it happen?
1) Platelets tend to be contaminated by skin contaminants from collection process
a) Scarred collection sites (>20 donations) can harbor organisms
b) Diversion pouches that accept the first 30-50 mL or so of collected blood in a donation are meant to harvest potentially contaminated skin plugs
2) RBCs more commonly contaminated by an organism growing in the donor’s blood (often asymptomatic)
a) Surprising amount of silent bacteremia is seen
b) Yersinia enterocolitica historically most common (large case series from CDC showed most bacteremic donors had mild GI symptoms before or after donation)
Organisms identified depend on product.
1) Red cells
a) Gram-negative rods (endotoxin-makers that like growing in cold temperatures):
• Yersinia enterocolitica (most common historically)
• E. coli
• Enterobacter/Pantoea sp
• Serratia marcescens and S. liquifaciens
• Pseudomonas species
b) Gram-positive cocci (much less commonly)
• Staph. Epidermidis
• Propionibacteria
• Staph aureus
2) Platelets
a) Vast majority are gram-positive cocci (skin contaminants including those listed above); majority result in only mild reactions (if at all)
b) Gram negative rods can also contaminate and are much more likely to cause fatalities than gram-positives (reported examples include Serratia, E. coli, and Klebsiella species)
3) Plasma products
a) Uncommonly contaminated
b) Few reports involving water bath contamination with Pseudomonas species
Transfusion-related acute lung injury (TRALI)
Currently the #1 cause of transfusion-related fatality in the US! (see pg 1)
1) Incidence varies widely: 1:1200 to 1:190,000 transfusions
Two almost identical standard definitions:
1) National Heart, Lung, and Blood Institute (NHLBI) Working Group and Canadian Consensus Conference Panel
a) New acute lung injury within 6 hours of a transfusion; ALI defined:
• Hypoxemia with PaO2/FiO2 < 300 mm Hg (or O2 sat <90%) and bilat CXR infiltrates
b) Lack of other risk factors for pulmonary edema
c) No pre-existing acute lung injury
2) Usually also with fever, chills, transient hypertension then hypotension
3) Platelets/plasma transfusions most often, but also with RBCs/whole blood
Clinical differential diagnosis:
1) ARDS: TRALI may look exactly like ARDS, but TRALI usually resolves in 24-48 hours.
2) Transfusion-associated circulatory overload (TACO): May be identical clinically, complete with a “wet” chest x-ray, but TRALI is usually associated with fever (unlike TACO) and does not respond to diuretics
3) Anaphylactic reactions (generally afebrile)
4) Acute pulmonary and myocardial disorders
Pathophysiology: Two currently accepted pathways that are very closely intertwined 1) The neutrophil is the villain in the pathophysiology of TRALI!
a) Regardless of the “pathway” taken (outlined below), secretion of toxic oxygen free radicals and other substances by the PMN damages pulmonary endothelial cells and capillaries and leads to vascular leakage and TRALI
b) This is merely an exaggeration of normal PMN activity in killing and neutralizing bacterial infections (esp. secretion of reactive oxygen species)
c) The lung is a HUGE sink for PMNs; almost 30% of body PMNs live in the lungs at all times!
2) Donor antibody pathway for TRALI
a) Anti-HLA or anti-neutrophil antibodies from the donor bind to antigens on the recipient neutrophils (see image)
b) Antibody-PMN complexes deposit in pulmonary vasculature and activate the PMN bactericidal response.
c) Secretion of toxic oxygen radicals and bactericidal enzymes leads to damage to pulmonary endothelial cells lining capillaries, with resultant leakage of fluids into the alveolar spaces (pulmonary edema).
d) This mechanism may also occur with recipient antibodies against donor WBCs, but this is less common.
e) Problem: This mechanism alone does not explain all demonstrated TRALI reactions, nor does it explain why TRALI does NOT occur in settings where it SHOULD (donors with antibodies against recipient antigens that do not have reactions at all)
3) “Two-event” pathway for TRALI
a) First event: Pre-transfusion condition that activates lung endothelial cells and primes PMNs (prepares them for microbicidal action)
• Examples include sepsis, major surgery, massive transfusion
• The activation puts the patient in a state where an additional “push” could start the process of
b) Second event: Transfusion of stored blood product (+/- antibodies)
• Stored blood products accumulate substances called “biologic response modifiers” (BRMs) that can prime/activate destructive neutrophils (bioactive lipids such as lyso-phosphatidylcholines or “lyso-PCs”, soluble CD40 ligand).
• Either BRMs or antibodies mentioned in the first hypothesis may induce capillary damage by activating the primed PMNs to secrete toxic substances
c) Combination of these events leads to capillary damage and subsequent pulmonary edema.
d) This mechanism helps explain the inconsistencies with the donor antibody pathway and synthesizes the two pathways nicely; still controversial though!
Diagnosis
1) Difficult, as it is often confused for something else
2) Typical early findings: bilat CXR infiltrates, oxygen sat <90%, no evidence of volume overload (no JVD, normal wedge pressure, normal BNP)
3) Lab findings may include demonstration of anti-HLA and/or anti-neutrophil
antibodies, and possibly increased biologic response modifiers in the bag.
a) Remember, this is a clinical and radiographic diagnosis; confirming the presence of donor antibodies may take days or weeks!
Treat with respiratory support (oxygen, maybe intubation).
1) Mortality reported between 5 and 25% 2) 80% recover quickly
- diuretics not indicated
Prevention
1) Current AABB mandate for transfusion centers to reduce TRALI risk
2) Implicated donors (with antibodies found) should be deferred from donation
3) Use of all (or mostly) male plasma has been shown to decrease the risk of TRALI (females have more anti-HLA and anti-neutrophil antibodies because of pregnancy).
4) Some centers have begun testing parous female PLT donors for anti-HLA +/- neutrophil antibodies and deferring those who have antibodies
5) Strategies only address antibody-formers and ignore two hit model
Possible TRALI (pTRALI)
- aka transfused ARDS
New ALI w/in 6 hrs of transfusion c a clear temporal relationship to an alternative risk factor for ALI
- incidence does not dec c male plasma or HLA/HNA antibody
- mortality rate similar to ARDS
Major risk factors: sepsis, pneumonia, aspiration, multiple fractures, pancreatitis
- minor risk factors: stroke, drowning, lung damage, amiodarone, high altitude
Acute reactions presenting without fever
Allergic reactions
Mild allergic (urticarial, cutaneous) transfusion rnxs
1) Very commonly reported reaction (1-3%)
2) Usually localized hives, but may have more severe swelling around eyes and lips (angioedema), mild respiratory symptoms, and laryngeal edema (see moderate reactions below)
Mechanism
a) Type I (IgE-mediated) hypersensitivity to transfused plasma proteins (not usually a specific, identifiable allergen)
b) Mast cell secretion of histamine and resultant cytokines and other mediators of allergic reactions
Prevention and treatment options
a) Diphenhydramine (Benadryl) IV 25-50 mg as treatment, may use PO form (same
dose) as pre-transfusion prophylaxis
• Probably not cost-effective or advisable for routine transfusions, but may be useful in those with a history of reactions
b) Washed products work too (not usually done)
c) May restart transfusion after hives clear.
Moderate allergic (anaphylactoid) transfusion reactions
1) Some allergic reactions fall between the two classic categories
2) May present with upper/lower airway obstruction +/– cutaneous manifestations
a) Upper airway:
• Stridor, hoarseness, “lump” in throat
b) Lower airway:
• Wheezing, chest tightness, dyspnea
3) Some of these patients may respond to IV diphenhydramine and not require
epinephrine, while others will need epinephrine
Differential diagnosis:
a) Acute HTR
• Typically febrile
• Most acute HTRs don’t present this early
• Must rule out nonetheless
b) Septic transfusion reaction
• High fevers and lack of skin findings in septic reactions may be only ways to
distinguish early
• If unclear, give epinephrine anyway
c) Acute hypotensive reactions
• These reactions (see below) have hypotension only, without respiratory or
skin findings
Confirmation
a) ALL patients with a severe allergic reaction should have, at minimum, a check of their pretransfusion sample for serum IgA levels
b) Those with very low/undetectable IgA levels (<0.05 mg/dL) should be tested for anti-IgA (detects IgG antibody but could predict the possibility of IgE)
Acute hypotensive reactions
Reactions that are similar to severe allergic reactions but ONLY have severe hypotension (no skin symptoms, no GI complaints, no respiratory issues)
1) CDC definition:
a) Over 30 mm Hg drop in systolic BP with diastolic < 80 mm Hg
b) Occurs less than 15 minutes after the start of transfusion
c) Resolves within 10 minutes after transfusion stopped
Classically associated with two situations (and often with both in the literature)
1) Patients taking angiotensin-converting enzyme inhibitors (ACEi)
a) Rapid onset of flushing and hypotension in transfused patients who are on ACEi
(e.g., Vasotec, Lotensin, Zestril, Capoten).
b) Probably caused by accumulation of increased bradykinin or bradykinin metabolites generated during storage
• ACE inhibitors prevent full metabolism of bradykinin (actually stops at a potent metabolite with a longer half-life)
• Bradykinin/metabolite causes marked but transient hypotension (bradykinin half-life is on the order of seconds, metabolite mentioned above longer but
still only minutes)
2) Patients receiving blood through negatively charged filters
a) Seen historically with certain negatively charged bedside leukoreduction filters
b) Also seen in therapeutic apheresis procedures such as LDL apheresis (due to interaction with negatively charged filters) and plasma exchange with albumin replacement (especially if on ACEi)
c) Most concerning, reported with reinfusion of intraoperative blood (cell saver) that is filtered before infusion
Transfusion-associated dyspnea (TAD)
A total garbage can diagnosis, in my view!
Definition:
1) Acute onset of respiratory distress less than 24 hours after transfusion
2) TRALI, TACO, and allergy ruled out
c. Should not be a common diagnosis!
Transfusion-associated circulatory overload (TACO)
Acute onset of congestive heart failure as a direct result of blood transfusion
1) Dyspnea, orthopnea, bilateral rales, with hypoxia
2) Systolic hypertension (widened pulse pressure), tachycardia, jugular venous distension, pedal edema, headache
3) Usually afebrile
4) X-rays with bilateral predominantly basilar infiltrates, widened cardiac silhouette
5) Proposed diagnostic criteria include some of the above signs/symptoms PLUS:
a) Hypoxemia (<90% saturation on room air)
b) Bilateral CXR infiltrates
c) Reaction occurring within 6 hours of transfusion
Patients most at risk (though any patient may get TACO if transfused rapidly):
1) Patients with pre-existing CHF
2) Very old (>85% occur in patients over age 60) and very young (to a lesser extent)
3) Renal failure
4) Chronic anemias (e.g., sickle cell, thallasemias), due to compensation for anemia with increased plasma volume
Differential diagnosis:
1) TRALI
a) May be clinically and radiographically identical (though cardiac silhouette widening may not be present in TRALI)
b) See distinctions below
2) Allergic/anaphylactic reactions
a) May present VERY early in transfusion (first few drops)
b) Not responsive to diuretics or positional changes
3) Coincidental cardiac or pulmonary issues unrelated to transfusion
a) Acute MI or pulmonary embolism
b) Valvular heart disease with decompensation
d. Distinguishing from TRALI
1) Clinical (response to diuretics/positional changes in TACO, fever in TRALI)
2) Lab: Elevated brain natriuretic peptide (BNP) suggests TACO (some use ratio of pre- to post-transfusion >1.5 AND elevated post-transfusion)
3) Finding anti-HLA and/or –HNA antibodies as above establishes TRALI
Acute Pain Reactions
a. Sudden onset pain in trunk/extremities
b. No predictable risk factors, no way to prevent
c. Lab workup is negative, and symptoms resolve shortly after transfusion
d. May require narcotics to relieve pain
Delayed reactions presenting with fever
Delayed hemolytic transfusion reactions (DHTRs)
Hemolysis occurring at least 24 hours but less than 28 days after transfusion (CDC definition, but rare reports up to 6 weeks).
Pathophysiologic possibilities
1) Anamnestic response
a) Mechanism:
• Patient exposed to non-ABO red cell antigen not present on own RBCs
• Antibody is formed but fades from detection over time (if patient is not retested after transfusion, which is common, antibody might never be known)
• Patient is re-exposed to antigen in a future transfusion (because antibody screen is negative for the offending antibody)
• Anamnestic rapid production of IgG antibody vs. target antigen
b) This mechanism is typical for Kidd, Duffy, Kell antibodies
2) Primary response
a) Mechanism:
• Patient exposed to non-ABO red cell antigen not present on own RBCs
• Antibody is formed quickly
• Antibody attacks still-circulating transfused red cells carrying target antigen
b) MUCH less common than anamnestic mechanism, but possible
Classically leads to extravascular hemolysis
1) IgG antibodies coat red cells and lead to their removal in the liver/spleen
2) Peripheral smear will commonly show spherocytes in this setting
3) NOTE: Delayed HTRs due to Kidd (Jk) antibodies may be intravascular and severe (these antibodies are capable of fixing complement).
Signs/symptoms
1) Often completely asymptomatic
2) Fever and anemia of unknown origin
3) Mild jaundice/scleral icterus may be seen
Transfusion-associated graft-vs-host disease (TA-GVHD)
A nearly-always fatal transfusion complication resulting from an attack on recipient
cells by viable T-lymphocytes in a transfused blood product
TA-GVHD sequence/requirements:
1) Viable, active T-lymphocytes are transfused.
a) Minimum number needed to cause TA-GVHD unknown.
2) Donor and recipient are not HLA-identical.
3) Recipient is unable to respond to neutralize the effect of the transfused WBCs.
The normal response:
1) Transfused T-lymphocytes (CD4, CD8, and NK cells) mount an immune response against foreign HLA host tissues (top left of figure 11 below).
2) Normally, host lymphs (CD8 and NK cells especially) counterattack and neutralize the response (top right of figure 11 below).
Lack of host neutralization (bottom right of figure 11 above) may lead to TA-GVHD, with continued T-lymphocyte attack on host tissues
1) Almost uniformly fatal, so thankfully rare
2) Patients present with:
a) Fever 7-10 days post-transfusion
b) Face/trunk rash that spreads to extremities
c) Mucositis, nausea/vomiting, watery diarrhea
d) Hepatitis
e) Pancytopenia and subsequent marrow aplasia
• This is what leads to the fatal consequences, with most patients dying from overwhelming infections
Patients at-risk for TA-GVHD:
1) Immunosuppressed patients
a) Congenital T-cell deficiencies (DiGeorge’s, SCID, Wiskott-Aldrich)
b) Stem cell or marrow transplant recipients
c) Patients taking chemo agents that attack T-cells (Fludarabine, purine analogs)
d) Aplastic anemia patients
e) Patients with solid tumors getting intensive chemotherapy/radiation
2) Intrauterine transfusions, premature neonatal transfusions, and neonatal exchange transfusions
3) Hematologic malignancies (esp. Hodgkin’s)
a) Hodgkins seems to have an inherent cellular immune defect that puts these patients at risk
b) Patients with other heme malignancies are more at risk due to intensive treatment regimens
4) Patients with solid tumors undergoing intensive treatment
5) Granulocyte transfusion recipients
a) Viable, fresh T-lymphocytes present in this short-shelf life product
6) Receiving blood from a first-degree relative donor or receiving HLA-matched units
a) Specific risk: HLA-heterozygous recipient from an HLA-homozygous donor
(“One-way HLA match”); see Figure 12 below
• Child 2 gets blood from child (child 1 is HLA homozygous, while child 2 shares one haplotype with child 1)
• Child 1 attacks and sees child 2 as “non-self” (due to the non-shared haplotype), but child 2 does NOT see child 1 as “non-self” due to the shared HLA haplotypes
b) As a result, there is an “attack” (transfused child 1 T-lymphs vs child 2 HLA bearing cells) but no “counterattack” by child 2 T-lymphs vs. child 1 T-lymphs.
c) This occurs most frequently in families, but also occurs in less HLA-diverse populations (most famously in Japan)
d) This interaction can lead to TA-GVHD in a completely immunocompetent recipient
Delayed reactions presenting without fever
Delayed Serologic Transfusion Reaction (DSTR)
Described above in the DHTR section
A new antibody in a recently transfused patient, without evidence of hemolysis
Post-transfusion Purpura (PTP)
Rare reaction, with marked thrombocytopenia and increased risk of bleeding about ten days following transfusion (may be below 10,000/μL)
1) Bleeding is often mucocutaneous (mouth and nose, GI tract); intracranial hemorrhage is possible but occurs in less than 10% of cases typically
2) Triggering transfusion platelets or RBCs (more commonly RBCs/whole blood)
3) RBC products contain substantial amounts of platelets and soluble platelet antigens
Multiparous females especially at risk (5:1 female-male ratio)
Caused by antibody vs common PLT antigen
1) Anti-HPA-1A (PLA1; present in 98%) most common culprit (70-80% of cases)
2) HPA-1A negative patients are exposed through pregnancy or transfusion.
3) Transfusion after antibody is formed leads to devastating destruction of platelets.
4) HPA-1A-positive transfused platelets and HPA-1a-negative patient platelets are both destroyed, which is weird, right?
a) Most likely because antibody has autoantibody activity
b) Passive adsorption of Ag/Ab complexes or soluble PLT Ags also suggested
Differential diagnosis is challenging and difficult
1) TTP, ITP, DIC, HIT all can share features
2) Even more difficult in patients already thrombocytopenic
Tx: IVIG reverses the process and normalizes platelet count in about 3-5 days
1) Due to this, plasma exchange is uncommon today (only if IVIG doesn’t work)
2) Mortality historically 10% without treatment; now near 0% with treatment.
Platelet transfusion should be avoided if possible (ineffective, may worsen?)
Future platelet transfusions should be negative for target antigen
Iron overload
a. Each unit of RBCs: 200-250 mg iron (generally, 1 mg iron per 1 mL RBCs)
b. Lifetime load of ~50-100 transfusions in 70 Kg person = risk for overload.
1) Hepatic, cardiac, endocrine organ, RE system deposition is especially damaging
2) May present with hepatic or cardiac failure, diabetes, thyroid abnormalities
3) Big risk in chronically transfused patients
c. Exchange transfusions reduce risk
d. Iron chelators (deferoxamine, deferiprone, deferasirox) may remove iron from hepatic stores and from RE system
Consequences of significant reactions
1. FDA requirements
a. If there is suspicion that a death is transfusion-related, FDA requires notification “as soon as possible” by phone, fax, or e-mail (formerly 24 hours, and most try to comply)
1) Official rule says “confirmed” death, but FDA cites facilities regularly for not reporting deaths that are “suspicious” for being caused by transfusion
2) It’s ok to report a death that is suspicious (even a little), then follow-up as below
b. Initial report with a full investigation and written report within 7 days
2. JCAHO
a. Acute hemolytic transfusion reactions are “sentinel events” and require intensive investigation (Root Cause Analysis) and reporting to JCAHO.


Additional testing for suspected hemolysis:
1) Repeat antibody screen (on both pre- and post-transfusion samples); consider different enhancement (PEG, LISS, cold/warm incubation, etc) or platform
2) Repeat crossmatch with pre- and post samples
a) If no serologic crossmatch was done (i.e., if computer crossmatch used), it should be done if there is suspicion on first-tier investigation
b) Best done with tube technique including immediate spin and IAT phase readings +/- 37 C reading (gel does not necessarily detect ABO incompatibility)
3) Elution studies if DAT is positive to determine specificity of the antibody
4) Haptoglobin
a) Haptoglobin binds to free hemoglobin molecules, facilitating their clearance from the circulation by monocytes and macrophages in the RE system
• This interaction prevents iron from escaping through the kidneys and lessens any toxic effect of hemoglobin in the kidneys
b) Levels decrease sharply in acute intravascular hemolysis (traditional belief; more current data suggests that it decreases in extravascular hemolysis as well)
c) Long turnaround time and acute phase reaction make for limited usefulness in acute setting.
• If you must use, compare pre- and post levels.
5) Direct and indirect bilirubin
a) Really more useful to confirm, not make diagnosis
b) Both will rise quickly, peak in less than 10 hours, may be normal within 24 hours (if liver is OK)
c) This is in contrast to traditional teaching that acute hemolysis does not elevate direct bilirubin
6) Lactate dehydrogenase (LDH)
a) Another marker of hemolysis, as LDH is abundant in RBCs (especially LD2 and LD1 isoenzymes)
b) Not specific for intravascular hemolysis (+/- in extravascular, too)
7) Urine hemoglobin
a) Not as sensitive or as fast as hemoglobinemia for intravascular hemolysis
b) Really only useful if testing for visible hemoglobin has been delayed or if there is doubt about a positive visible hemoglobin check
c) Remember that hematuria does not equal hemoglobinuria!


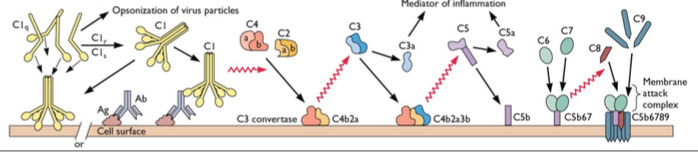

Net effects on various systems in AHTRs:
a) Inflammatory consequences:
TNF-α, IL-1β, and IL-6 strongly promote fever
• WBCs activated and stimulated by all of above as well as IL-8 and CCL-5
b) Coagulation consequences:
• Direct intrinsic path activation by Ag-Ab complex interaction with factor XI
Indirect activation of extrinsic path by TNF-α stimulation of tissue factor
• Decreased protein C inhibition via decreased thrombomodulin
• Tissue factor activation also predisposes to DIC (10% of patients)
c) Circulatory consequences:
• Increased C3a/5a, IL-1β, and TNF-α stimulate increased nitric oxide (NO) levels, which leads to systemic vasodilation
• Bradykinin generation from antigen-antibody complexes likewise promotes transient systemic hypotension
• On the other hand, massive free hemoglobin release may reduce local NO, leading to at least localized vasoconstriction
d) Renal consequences:
• Sympathetic response to hypotension leads to renal vasoconstriction
• Free hemoglobin scavenges renal NO, promoting vasoconstriction
• Renal microthrombi from diffuse coagulation also decrease renal blood flow
• Hemoglobin-free RBC stroma also damages renal tubules directly
• All of above contribute to risk for acute tubular necrosis, with resultant
oliguric renal failure in about 1/3 of confirmed acute HTRs
e) Respiratory consequences:
• Anaphylatoxins promote bronchoconstriction from histamine release, with resultant wheezing/dyspnea
• Aggressive hydration during resuscitation gives pulmonary edema risk

FNHTRs
Lab findings
1) None; negative hemolysis workup (diagnosis of exclusion)
Treatment
1) Antipyretics (acetaminophen)
2) Meperidine (Demerol) for more severe chills; use with caution!
Prevention
1) Acetaminophen premedication may prevent fever, but is not reliable
2) Preventing FNH during RBC transfusions
a) Most are due to post-transfusion WBC production of pyrogenic cytokines.
b) Leukocyte reduction works extremely well to prevent vast majority of these reactions; nearly all blood banks leukoreduce using pre-storage timing (during or very shortly after collection)
3) Preventing FNH during platelet transfusions
a) Most are due to pre-transfusion cytokine production by WBCs
b) Bedside leukoreduction is ineffective; substances are already in the bag!
c) Pre-storage leukoreduction best, but reactions in ~0.1 to 1% of PLT transfusions
• Exception: Soluble CD40 ligand is PLT-derived and causes fever, so LR wouldn’t prevent reactions caused by sCD40L (minority)

Transfusion-related sepsis
Signs/symptoms
1) Earlier symptoms seen in more severe reactions and more often with RBC transfusions (may occur within the first few minutes of transfusion; see fig 7)
2) Rapid onset high fever (often greater than 4oF/2C)
3) Rigors (true shaking chills with rigidity)
4) Abdominal cramping, nausea/vomiting
5) Hypotension/shock
6) DIC
Differential diagnosis:
1) Acute HTR (always!); Severe septic reactions, however, usually are more “acute” and dramatic in presentation than the typical AHTR
2) Anaphylactic transfusion reaction: Can also be dramatic and very early in the transfusion. Usually NOT febrile.
3) Febrile nonhemolytic transfusion reaction (FNHTR): Milder septic reactions and many PLT contaminations can overlap with FNHTR significantly (as a result, many culture units as part of FNHTR workup protocol)
4) Sepsis from non-transfusion source (infected lines and/or fluids, coincidental presentation)
Lab findings
1) Discolored RBC product (+/-); contaminated RBCs may turn DARK or purple
2) May have hemoglobinemia/uria (non-immune)
3) DAT negative (unless coincidental)
4) Gram stain positive in only half to 2/3 of proven cases!
a) Source of the gram stain and culture is very important
b) Avoid culturing or staining a segment (unless nothing else is available); false
negatives when culturing segs only have been described
c) Remember to also culture associated IV fluids and consider that an indwelling IV catheter may be a source of contamination
5) Culture is proof positive (when same organism is cultured from both unit and recipient; even better if from the donor as well!)
Treatment
1) Immediate IV antibiotics; treat presumptively with broad spectrum coverage, then adjust as necessary
2) Pressure/respiratory/general support as needed
3) Don’t forget: Quarantine all other products from the same donation if a reaction suspicious for sepsis occurs! Notify blood collection agencies promptly!
Prevention
1) Careful donor history
2) Proper phlebotomy technique; use of diversion pouches (mandatory now), strict attention to possible site contaminants
3) Leukocyte reduction filters may decrease risk (decrease in Yersinia concentration)
4) Routine detection of platelet contamination required by AABB Standard
a) Culture-based methods (culture taken 24 hours after collection):
• BacT/ALERT system: Microbiology standard blood culture equipment that is most commonly used method; detects decrease in pH from production of CO2 by growing organisms
• Pall enhanced bacterial detection system (eBDS): Detects bacterial use of oxygen
b) Pre-issue methods (performed shortly before issue)
- Neither approved system is widely used as of this writing
- VeraxPGD system (approved for detection in leukocyte reduced apheresis PLTs and pooled whole blood derived PLTs); detects bacterial cell wall antigens
• Recent report: approximately 1 in 3000 culture-negative apheresis PLTS are Verax-positive before issue (confirmed by second culture of unit)
• Problem: Cost is direct to transfusion service; resistance
BacTx system (approved only for pooled whole blood derived PLTs)
5) Despite detection methods, false negatives occur, and pathogen reduction may be the ultimate answer



Severe allergic (anaphylactic) transfusion reactions
1) Opposite end of hypersensitivity reaction spectrum
2) Uncommon (1:20,000 to 50,000 transfusions)
Presentation
a) Anaphylactic shock very early in the transfusion
b) Acute hypotension, lower airway obstruction, abdominal distress, systemic crash
c) Virtually all of these patients have skin findings (urticaria, angioedema, generalized pruritis)
What’s the allergen?
a) Classic history: IgA deficient recipient who has formed an anti-IgA of the IgE class (which induces a severe type I hypersensitivity reaction)
• Seen only in those with undetectable IgA levels (lower than what the allergists define as IgA deficient; i.e. IgA < 0.05 mg/dL)
• Problem: It’s easy to detect IgG anti-IgA, but REALLY hard to detect IgE anti-IgA!
• Vast minority of patients with anaphylactic-type reactions have demonstrable IgA deficiency with detectable anti-IgA
b) Haptoglobin deficiency in Asian patients; anti-haptoglobin in a recipient can give same type of severe reaction as anti-IgA
c) Latex, drugs, foods in donors can lead to severe reactions in susceptible recipients (recent report of patient with nut allergy having anaphylaxis after
plasma transfusion from donor who ingested peanuts before donation)
d) Scattered reports of donors with IgE antibodies transmitting a temporary hypersensitivity to the recipient, with resultant severe allergic reactions
e) BOTTOM LINE: While all of the above are possible, it is very uncommon to find a specific reason why a patient suffered a severe allergic reaction
Allergic reactions
Prevention
a) IgA-deficient (IgAD) products traditionally given for those with demonstrated antibodies, but sometimes given for those with low IgA alone
• IgAD options: Washed cellular products (RBCs, PLTs), or products from IgA-deficient donors (All products)
• If possible for future transfusions, may also back autologous units
b) Washed cellular products for those with demonstrated severe allergic reactions
and no demonstrable IgA deficiency
• While this is fairly common practice, MOST patients with history of severe
allergic reactions can get future untreated transfusions without harm (they
should be monitored VERY carefully however!)
c) Benadryl probably insufficient by itself for prevention or treatment; if necessary, may use corticosteroids +/- additional histamine blockers
Treatment
a) Epinephrine immediately (0.2-0.5 ml of 1:1000 IM/SQ)
b) SQ or IM preferred, but may give IV if already crashed.
Acute hypotensive reactions
Diagnosis
1) Clearly a diagnosis of exclusion
2) Rule out:
a) Acute HTR by workup and lack of fever
b) Severe allergic reaction by lack of skin and respiratory findings (as well as transient nature of process)
c) Septic reaction by lack of high fever and other clinical findings (GI complaints, transient nature of process)
Management
1) STOP the transfusion! (short half life of bradykinin leads to rapid resolution)
2) Give fluids, consider epinephrine if not resolved promptly
Prevention
1) No routine prophylactic measures necessary
2) Avoid bedside leukoreduction filters (not really a problem in most places, as most leukocyte reduction is done in blood centers or transfusion services)
3) Stop ACEi before therapeutic apheresis procedures
TACO
Treatment
1) Stop the transfusion, evaluate, sit patient up
2) Give supplemental oxygen
3) Diuretics to decrease blood volume
4) In severe cases, therapeutic phlebotomy may be indicated
Prevention in at-risk patients
1) Control infusion rates (1 mL/Kg/hour).
2) Split units into aliquots when possible.
3) Consider lower volume units (using CPD-RBCs rather than AS-RBCs, for example)
or volume reduction of certain products.
DHTR
Lab findings
1) Icteric serum
2) DAT positive (classically “mixed field”)
3) Anemia
4) Newly identified red cell antibody
5) Spherocytes on peripheral smear
6) Elevated LDH and indirect (and often direct) bilirubin, decreased haptoglobin (even if hemolysis is extravascular)
Treatment
1) As for AHTR if severe and intravascular
2) Often no treatment necessary
g. DHTR vs. “delayed serologic transfusion reaction” (DSTR)
1) These two terms used interchangeably historically (has led to some confusion)
2) Official definition of DSTR:
a) The presence of a new, clinically significant red cell antibody in a patient
transfused > 24 hours and < 28 days ago without evidence of that antibody,
AND:
b) Complete lack of evidence of hemolysis after careful evaluation
3) Don’t diagnose DSTR without studying the patient carefully (repeat antibody screen on pretransfusion sample if possible; evaluate bilirubin, haptoglobin, LDH, peripheral smear, etc).
a) Any evidence of hemolysis in study above changes the diagnosis from DSTR to DHTR

Radiation is used to deactivate the T-lymphocytes in transfused blood products
1) 2500 cGy (“rad”) dose required targeted to center of bag, with at least 1500 cGy in all parts of the bag
2) Irradiation dose deactivates lymphocytes without significantly damaging anything else (including PMNs).
3) Why not leukocyte reduction?
a) Minimum threshold for TA-GVHD prevention is not known
b) Case reports of TA-GVHD from leukoreduced units
Patients probably NOT at risk (but often get irradiated products anyway).
1) Solid organ transplant recipients
2) Term neonates
3) AIDS patients (CD8 cells that counterattack preserve function until late in disease).
4) Patients receiving previously frozen plasma products (FFP, cryoprecipitate)
a) Frozen/thawed/deglycerolized RBCs should be irradiated if recipient is at risk for TA-GVHD; since they were cryopreserved, viable T-lymphs can survive.
Don’t use irradiation for:
1) Preventing CMV transmission (leukocyte reduction)
2) Peripheral progenitor cell infusions (think about it)
i. Gamma irradiation (stand-alone units, usually in transfusion service) and x-ray
irradiation (commonly performed in radiology or radiation oncology areas) are used
interchangeably and are equally effective
j. Maximum storage: 28 days after irradiation or regular expiration date, whichever comes first
1) K+ and free hemoglobin increase in plasma
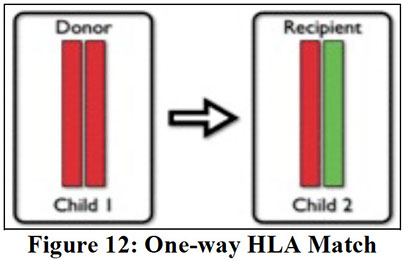
References
1. Blood Bank Guy. http://www.bbguy.org