
Lymph Nodes
Reactive Nonspecific Changes
Immunodeficiency-Associated Lymphoproliferations
Reactive Follicular Hyperplasia (RFH)
Reactive Paracortical Hyperplasia (RPH)
Infectious Causes of Lymphadenitis
Overview of Immunodeficiency-Associated Lymphoproliferative Disorders
Autoimmune Lymphoproliferative Syndrome
Immunomodulating Agent-Associated Lymphoproliferative Disorder
Posttransplant Lymphoproliferative Disorder, Early Lesions and Polymorphic
Posttransplant Lymphoproliferative Disorder, Monomorphic
Chronic Granulomatous Lymphadenitis
Suppurative Lymphadenitis
Mycobacterium tuberculosis Lymphadenitis
Atypical Mycobacterial Lymphadenitis
Mycobacterial Spindle Cell Pseudotumor
Cat-Scratch Disease
Bacillary Angiomatosis
Lymphogranuloma Venereum Lymphadenitis
Whipple Disease
Syphalitic Lymphadenitis
EBV+ B-cell Lymphoproliferative Disorders
- Infectious Mononucleosis
Histoplasma Lymphadenitis
Cryptococcus Lymphadenitis
Toxoplasma Lymphadenitis
Coccidioides Lymphadenitis
Herpes Simplex Lymphadenitis
Cytomegalovirus Lymphadenitis
Human Immunodeficiency Virus Lymphadenitis
Nonhematopoietic Proliferations in Lymph Node
Epithelial Inclusions in Lymph Node
Nevus Cell Inclusions in Lymph Node
Vascular Transformation of Lymph Node Sinuses
Angiomyamatous Hamartoma
Palisaded Myofibroblastoma
Metastatic Kaposi Sarcoma
Reactive Lymphadenopathies
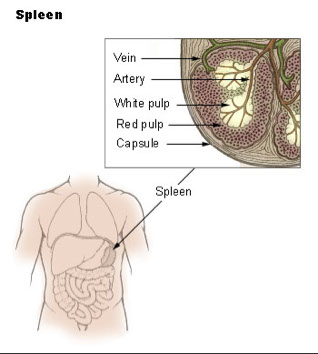
Spleen
Inflammatory Pseudotumor of Lymph Node
Progressive Transformation of Germinal Centers (PTGC)
Kikuchi-Fujimoto Disease
Rosai-Dorfman Disease
Kimura Disease
Unicentric Hyaline Vascular Variant Castleman Disease
Multicentric Castleman Disease
Rheumatoid Arthritis-Related Lymphadenopathy
Sarcoid Lymphadenopathy
Dermatopathic Lymphadenopathy
Hemophagocytic Lymphohistiocytosis
Lymphadenopathy Associated with Joint Protheses
Lipid-Associated Lymphadenopathy
Lymphadenopathy Secondary to Drug-Induced Hypersensitivity Syndrome
IgG4-Related Disease
Embryology, Anatomy and Histology
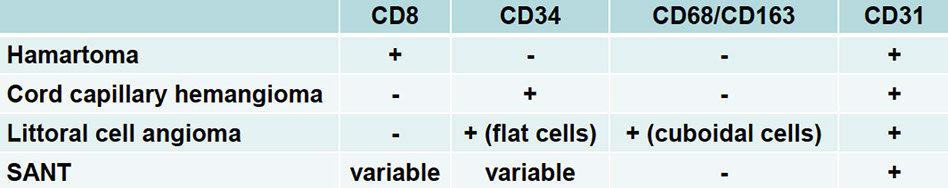
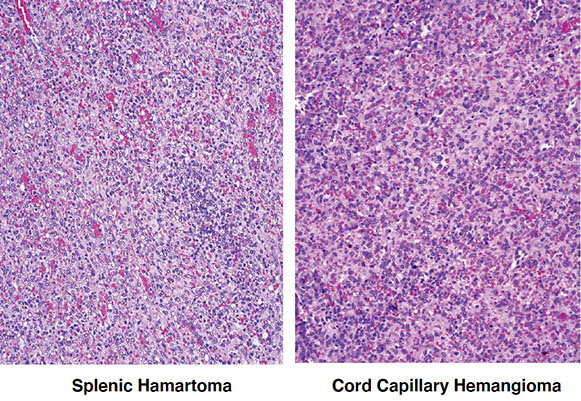
Splenic hamartoma
Cord capillary hemangioma
Sclerosing nodular angiomatoid transformation (SANT)
Littoral cell angioma
Splenic Marginal Zone Lymphoma (SMZL)
Splenic Diffuse Red Pulp Small B-cell Lymphoma
Diffuse Large B-cell Lymphoma (DLBCL) Arising in the spleen
Myeloid neoplasms
Lymph Nodes
Note: Primary lymphoid organs = BM and thymus
- site of lymphocyte birth and maturation by a process that does not involve antigens; have no follicles and make mature naive lymphocytes
Secondary lymphoid organ (LNs, spleen, MALT) activate naive lymphocytes with antigen and have follicles
Embryology
Content body
Anatomy, Histology
Lymph nodes are secondary lymphoid organs (they activate mature naive lymphocytes with antigen and have follicles)
- contain lots of afferent but few efferent lymphatics
Cortex is right under the capsule and has follicles with B cells; outer cortex has B cells and inner (deep) cortex has CD4+ T cells
- primary follicles are dense and dormant (lacks mantle and germinal center)
- secondary follicles pale and active c germinal center (where activated B cells proliferate after T cell activation) and mantle (with follicular dendritic cells [FDCs] that interact c mature B cells)
Germinal centers in secondary follicles are normally polarized c a dark centroblast region c blast-like B cells and a centrocyte region that is lighter c B cells c irregular nuclear contours and cleaved nuclei
- centroblasts and centrocytes both express CD10
Medulla = medullary cords (plasma cells and lymphocytes) and medullary sinuses (macrophages + reticular cells; communicates c efferent lymphatics)
- near the hilum; has lots of BVs and lymph sinuses
Paracortex bwt outer cortex and inner medulla has (CD4+) T cells (after undergoing differentiation in thymic cortex and medulla) and interdigitating cells (IDCs; express CD1a and S-100; antigen-presenting cells that help initiate immune response)
- has high endothelial (post-capillary) venules, where lymphs enter LN from blood
- atrophy of paracortex in DiGeorge syndrome bc do not have T cells
***T cells found in structures that begin with "P" = PALS in spleen and Paracortex of LNs; B cells found in follicles - follicles of cortex in LNs and follicles of white pulp in spleen***

[3]

Reactive Nonspecific Changes
Germinal Center polarization
- has dark zone (centroblasts) and light zone (centrocytes)
Cortex with B-cells, primary and secondary follicles
Paracortex c T-cells, blood vessels, interfollicular zone
Medullary cords have plasma cells and T cells
Sinuses c monocytes

B-cells in follicles and sprinkled in interfollicular region
(+) parafollcular immunoblasts
Mostly positive in T-cells in paracortex
CD4:CD8 ratio usually 3-4:1
Reactive Follicular Hyperplasia (RFH)
B9, Nonspecific, Hyperplastic follicles w secondary germinal centers
- clinically is a big LN, may be assoc c fever, fatigue
- pt age and evolution of dz important, as w size and location
Causes include medications, bacteria, viruses (EBV, HIV), autoimmune dz, Castleman dz, progressively transformed GCs, HIV-assoc lymphadenopathy, Rheumatoid lymphadenopathy (polytypic plasma cells and neuts in sinuses), and Syphilitic lymphadenopathy (thick capsule c plasma cells), usually cannot identify etiology
Location may suggest underlying dz
- cervical --> infectious mononucleosis
- posterior cervical --> Toxoplasmosis
- Parotid, submaxillary, epitrochlear --> HIV infx
- Cervical and axillary --> Cat-scratch dz
- Inguinal --> STD
- Supraclavicular --> Malignancy, esp in older pts
Gross: Generally, LNs >1 cm are abnormal
- inguinal LNs should be < 1.5 cm, epitrochlear <0,5 c
Micro: many large follicles, variety of size / shape
- have central germinal center (GC) and well-demarcated mantle zone
- occasional coalescence of follicles ok
- in spleen, reactive follicles are in the white pulp, in LN they are usually around the cortex, not medulla
Germinal center (GC): centroblasts and centrocytes, few T-cell, scattered histiocytes
- usually are polarized (have light and dark zones)
-- dark zone: many centroblasts (with high N:C ratio) and mits
-- light zone: predominantly centrocytes and follicular dendritic cells
Centroblasts: 3-4x size of small lymphs; have 1-3 nucleoli
Mantle zone: concentric layers of small naive (non-antigen exposed) B-cells
- proliferate to become centroblasts when exposed to antigen
IHC: GC and mantle zone express polytypic Igs and pan-B-markers
- centroblats and centrocytes CD10+, BCL6+, BCL2-
- T-follicular helper cells (Tfh) CD4+, CD10+, Bcl-6+, CXCL13+, PD1/CD279+
- Follicular dendritic cells (have 2 square-shaped adjacent nuclei) CD21+, CD23+, CD35+
- Histiocytes: CD68+, CD163+
May be associated with an EBV+ Lymphoproliferative disorder (EBV+ LPD)
- tend to occur early after transplantation
- overall architecture preserved with minimal interfolllicualr expansion
- Distribution of EBER+ cells often confined to a few follicles
- EBV+ T-cell hyperplasias also exist
- Dasatinib-related hyperplasia is another thing
Lymphoid "trafficking" can be seen in conditions with increased circuliating lymphocyts, eg viral, activated lymphcytes, immunoblasts also present
- if cytologically atypical, leukemia / lymphoma should be considered
Genes: Polyclonal IGH rearrangements
- No evidence of t(14;18) or IGH-BCL2 fusion sequences
DDx: Follicular lymphoma (FL), has polymorphic cells, inc mits (dec in FL), normal macrophages (dec in FL), neg Bcl-2 and t(14;18) (pos in FL), preserved architecture, GC morphologic variation, and no back to back follicules (back to back in FL)
- Atypical follicular hyperplasia - term for follicular lesions with some features suggestive of FL, now an uncommon diagnosis
- PTGC - usually assoc c RFH, PTGC has larger follicules than RFH, reactive centrocytes and centroblasts are BCL6+ and BCL2-, small round lymphocytes are BCL6- and BCL2+
- NLPHL - nodules are much larger than follicles, predominance of small lymphs (mostly reactive B-cells); LP cells are CD20+, CD45+, BCL2-; CD57 cells are increased in nodules and can form rosettes around LP cells
- LRCHL, nodular variant - nodular variant can closely resemble NLPHL, , neoplastic cells are CD15+, CD30+, CD20-/+, PAX5+, CD45-
- autoimmune or connective tissue disease (rheumatoid arthritis, Sjogrens)
- other infectious lymphadenitis (Toxoplasma gondii, syphilis, early viral [EBV or CMV])
- Castleman's disease
- May be nonspecific


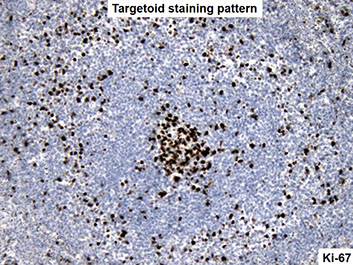
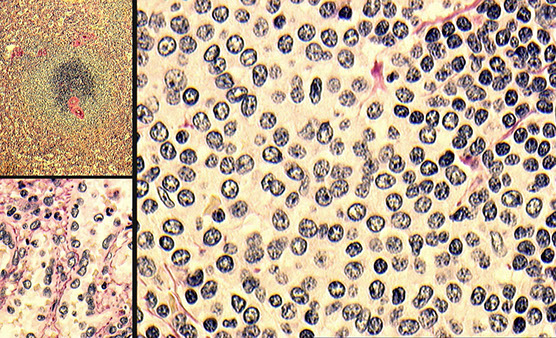
Reactive follicle - mantle zone is more attenuated near area of dark zone because that is where the naive B-cells have been recruited to become centroblasts in the dark zone. As the centroblasts undergo somatic hypermutation to match with antigen they mature into centrocytes (although many centroblasts undergo apoptosis); other cells include follicular dendritic cells (involved with antigen presentation) and tingible body macrophages (histiocytes; have variable apoptotic debris in them)

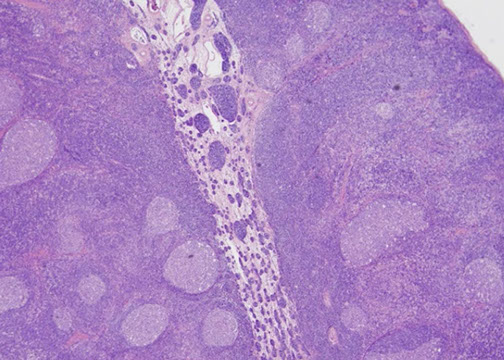

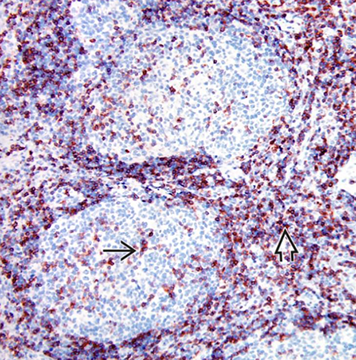
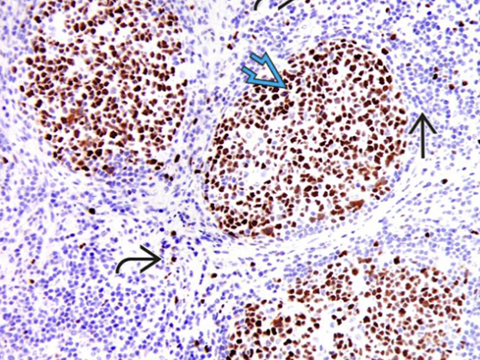





Reactive follicular hyperplasia (arrow to focal paracortical hyperplasia) [2]
Lymphoid "trafficking" can be seen in conditions with increased circuliating lymphocyts, eg viral, activated lymphcytes, immunoblasts also present
- if cytologically atypical, leukemia / lymphoma should be considered
CD3 in reactive follicles. Immunohistochemical stain for the pan T-cell marker CD3 highlights scattered small lymphocytes within germinal centers (solid black line). Many more CD3(+) lymphocytes are noted in the interfollicular region .(open black line) [2]
Immunohistochemical stain for Bcl-6 highlights centrocytes and centroblasts in the reactive germinal centers (open blue arrow) Mantle zone lymphocytes (straight black arrow) are negative; rare or scattered interfollicular lymphocytes (curved black arrow) are positive for Bcl-6. - Ki67 looks similar
Reactive hyperplasia, gross. The gross features mimic the microscopic features. Has variegated appearance (a malignant LN has a monotonous appearance) with mottled appearance showing red and tan areas. Central area of scar-like appearance is the hilum.
Reactive Follicular Hyperplasia (RFH). Follicles (B-cell compartment) have varying size and are well spaced from one another. There is a nice capsule and subcapsular sinus. In medullary compartment where plasma cells and memory B-cell live, the sinuses are open and patent. Note the relative abundance of the paracortex compared to the cortex, which is an important feature for B9 LNs.
In follicular lymphoma follicle with monotonous cellular composition, loss of the lymphoid cuff and no starry sky pattern.
In follicular hyperplasia has sharply circumscribed germinal center, starry sky pattern and nice lymphoid cuff
Hyperplastic lymphoid follicle. Germinal center with sharply demarcated mantle zone (solid blue arrow). Dark zone of GC with starry-sky pattern (open blue arrow) and light zone (open black arrow) with small centrocyte predominance [2]
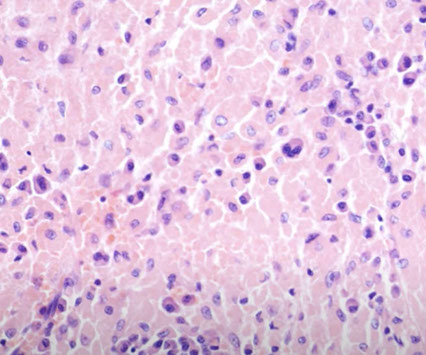
High magnification of GC with centroblasts (solid blue line) mixed with histiocytes with clear cytoplasm with nuclear fragments (open black line) and frequent mitoses (curved black line)
1 - 10
<
>

PD1 expression in RFH in a teenager with drug hypersensitivity. Stronger staining in reactive residual germinal center and weaker and more variable staining in reactive paracortical T-cells

Reactive Paracortical Hyperplasia
Expansion of paracortex from T-cells proliferation, which can be due to drugs, viruses and vaccines
- architecture is preserved
- Immunoblasts in a sea of small T cells and histiocytes (immunoblasts can be CD30+, but this is not Hodgkins!!)
- assoc c viral infx, post-vaccine lymphadenitis, Infectious mononucleosis (EBV), drug-induced hypersensitivity, dermatopathic lymphadenitis, histiocytic necrotizing lymphadenitis, SLE
Micro: T-cells appear monomorphous, small and mature appearing
- have almost evenly-spaced scattered interdigitating dendritic cells sprinkled throughout that gives it a mottled-effect
Reactive paracortical and reactive follicular hyperplasia tend to co-exist
DDx: may be nonspecific
- dermatopathic lymphadenitis
- viral lymphadenitis (CMV, HSV, EBV)
- drug-induced (phenytoin)
- Kikuchi-Fujimoto
- may be associated with a varying degree of an immunoblastic reaction
- Peripheral T-cell lypmhoma (PTCL) - some PTCL aberrantly express CD20, rarely CD79a, CD15, the may have aberrant subset antigens (double loss of CD4 and CD8, or double expression of CD4 and CD8), and antigen loss (CD7 not specific, CD5 negative in gamma delta, other PTCL)

Reactive paracortical hyperplasia [3]. Hyperplasia of the paracortex outside of follicles. Can have vaguely nodular areas of enlargement of the paracortex. The T-cells appear monomorphous, small and mature appearing. Have almost evenly-spaced scattered interdigitating dendritic cells sprinkled throughout that gives it a mottled-effect

Sinus Pattern
Includes sinus histiocytosis, Langerhans cell histiocytosis, Rosai-Dorfman dz, Hemophagocytic syndrome, and vascularization of sinuses
Crystal-storing histiocytosis, filled with kappa light chain from plasma cells here [7]
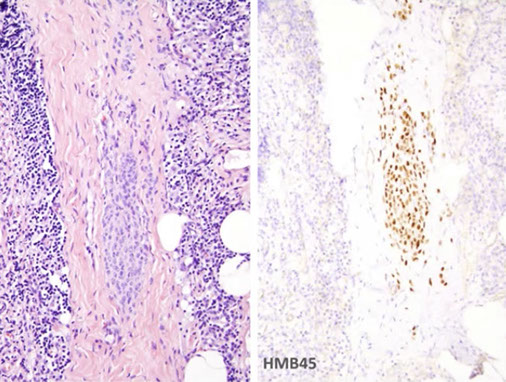
Lymph node nevus
Positive for HMB45, but cells are not atypical
Nodal nevus
Vascular Changes in lymph nodes
Prominent hilar vessels
- "Vascular transformation of the sinuses" - usually assocaited with blockage of efferent lymphatics (or veins) resulting in proliferation of hilar vessels
Plump high-endothelial venules
- Angioimmunoblastic T-cell lymphoma
Increased vascular elements
- Angioimmunoblastic T-cell lymphoma
- Bacillary angiomatosis
- Less commonly B-cell lymphoma
Vascular hyalinization
- inflammatory conditions
- hyaline-vascular Castleman disease
Vascular neoplasms
- hemangioma
- angiomyolipoma
- Kaposi sarcoma


Kaposi sarcoma
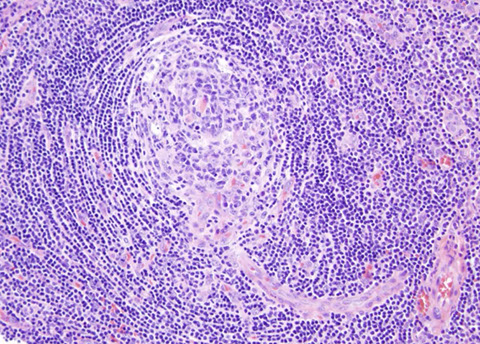
Castleman disease
Vascular transformation of the sinuses
Bacillary angiomaosis
1 - 4
<
>
Monocytoid B-cells
Reactive conditions
- Viral lymphadenitis
-- EBV, CMV, HIV / AIDS
- Toxoplasma lymphadenitis
Malignancy
- Marginal zone lymphoma
- Lymphoplasmacytic lymphoma (LPL)
- Mimicking monocytoid B-cells: certain T-cell lymphomas (AITL)
Monocytoid B-cells

Infectious Causes of Lymphadenitis
Chronic Granulomatous Lymphadenitis
-
Suppurative Lymphadenitis
-
Mycobacterium tuberculosis Lymphadenitis
-
Atypical Mycobacterial Lymphadenitis
-
Mycobacterial Spindle Cell Pseudotumor
-
Cat-scratch disease
Caused by Bartonella henselae; involves skin (red papule that can become encrusted) and Cervical and axillary LNs
- LNs have different micro characteristics depending on time frame
Dx: can confirm c serology
Micro: stellate necrotizing granulomas, neuts and B-cell hyperplasia
- clumps of bacili with Warthin-Starry or Hopps stain in walls of capillaries, in macrophages lining sinuses or near germinal centers and in areas of necrosis
- can also ID the bugs c PCR
Early - follicular hyperplasia and lots of histiocytes
Intermediate - granulomatous changes
Late - abscesses of various sizes c stellate necrosis and neutros surrounded by pallisading histiocytes
- sinuses filled c monocytoid B cells (unlike toxoplasmosis, peri-/intrafollicular epithelioid cells are absent)
DDx:
Px: self-limiting; resolves spontaneously
Cat-scratch disease

Bacillary Angiomatosis
-
Lymphogranuloma Venereum Lymphadenitis
-
Whipple Disease
-
Syphalitic (luetic) Lymphadenitis
Infection with Treponema pallidum
- can involve a variety of sites, such as lip, mouth, soft palate, tongue, rectum, anus
- usually has a prominent plasma cell infiltrate, but can have just lymphocytes or eosinophils
- shows both follicular and paracortical hyperplasia
Stage of infection corresponds to predominant histiology
- Primary: acute necrotizing lymphadenitis / chancre (rarely seen)
- Secondary: follicular hyperplasia with plasmacytosis, granulomas, perilymphadenitis (most commonly seen)
- Tertiary: necrotizing granulomatous lymphadenitis / gumma (rarely seen)
Lymphadenitis usually regional to the chancre
- typically inguinal, but could involve cervical nodes if infection via oral route
Exposure to sexually transmitted infection often not known or elicited
On physical exam, chancre may be misdiagnosed as another type of ulcer, related to cancer or other infection (HSV)
Micro: Follicular hyperplasia with inter- and intrafollicular plasmacytosis
- capsular and trabecular fibrosis with lymphoplasmacytic infiltrate
- paracortical expansion by clusters of epithelioid histiocytes (+/- suppurative features)
- endarteritis or venulitis may be present
IHC: anti-Treponema IHC can react with other spirochetes and requires serologic confirmation
DDx: immune disorders with plasmacytosis
- lymph nodes: RA, IgG4-related disease, Castleman disease --> although all of these diseases lack granulomatous component with associated neutrophils
- Extranodal sites: B-cell lymphoma with plasmacytic differentiation (eg MALToma) or carcinoma -- unusual pattern of inflammation, resolve with immunohistochemistry






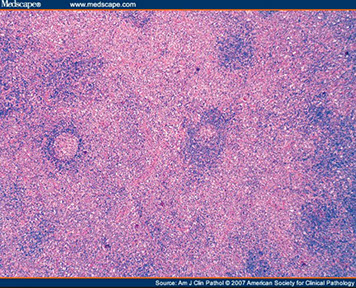
Syphalitic lymphadenitis [3]. Florid follicular hyperplasia with back-to-back appearance with little paracortex (may raise suspicion for malignancy), although the follicles have some degree of polarization. The capsule is intact, and appears thickened. There is some variability in the size and shape of the follicles (though that is a soft sign because can be seen in some low-grade follicular lymphomas). Plasma cells in the follicles in reactive GCs and in the thickened capsule.
Syphilis can have marked plasmacytosis, which is polytypic, can even have high IgG4 to IgG ratio, but IHC should reveal the organisms [3]
Syphilis. Areas with markedly-expanded paracortex with mix of lymphocytes, histiocytes, neutrophils, and sclerosis.
Also syphilis [3]
The anti-Treponema IHC can react with other spirochetes and requires serologic confirmation
1 - 6
<
>
EBV+ Lymphoproliferative Disorders
Ebstein-Barr Virus (HHV-4)
- double-stranded DNA virus isolated from Burkitt lymphoma cells
- EBV established lytic and latency programs in host cells
- EBV-infected B-cells persist within the memory of B-cell pool of most immunocompetent hosts
- EBV-encoded latent genes induce B-cell transformation by altering gene transcription and activting signaling pathways
- Immunosuppressed patients are at increased risk of developing EBV-associated B-cell lymphoproliferative disorders [8]
EBV+ Lymphoproliferative Disorders
- Spectrum from self-limiting hyperplasias to aggressive lymphomas
- Most often occur in immunodeficient patients; PTLD is the best studied
- Spectrum less well understood in T and NK cell proliferations
- Treatment options must be adapted to the immunodefiency context with an incremental approach to clinical management [8]
EBV+ B-cell Hyperplasias
WHO 2008: "Early Lesion", WHO 2016: "Non-destructive lesion"
- Mass-forming lesions with preservation of tissue architecture
- Clinical context is important to separate EBV+ hyperplasia from other non-specific causes of hyperplasia
- Polytypic kappa and lambda expression
- Occasional clonal IG or simple karyotypes present; although not indicative of malignancy, may lead to overdiagnosis as lymphoma
- Most hyperplasias regress spontaneously
- Rare concurrent or subsequent EBV+ B-LPD may occur; at least a subset are clonally related
Three kinds of EBV+ B-cell hyperplasias:
1. IM-like hyperplasia
2. Follicular hyperplasia
3. Plasmacytic hyperplasia
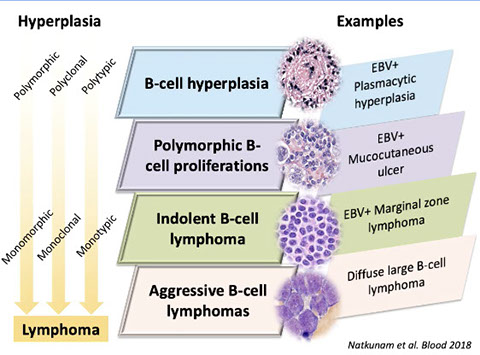


EBV+ LPDs.Similar spectrum of disorders can be seen in immunocompetent patients [8]
Genetic complexity - ordered progression of increasing karyotypic abnormalities with increasingly aggressive lesions [8] from Vakiani et al. Hum Pathol 2007
3 different kinds of EBV+ B-cell hyperplasias
3 - 3
<
>

Infectious mono can look like DLBCL
http://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(09)70351-1/fulltext

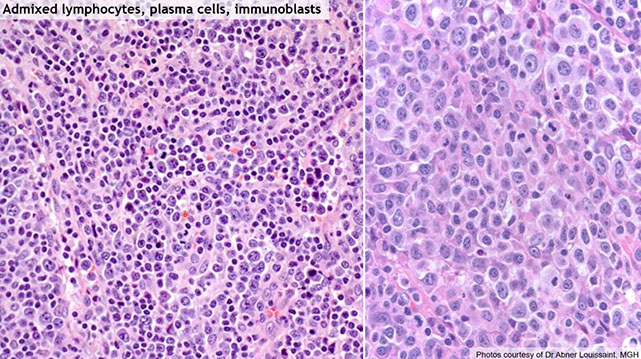
Infectious Mononucleosis (IM)
Paracortical hyperplasia, paracortex with moth-eaten appearance and has lots of lymphs which can be small to large sized, as well as immunoblasts, histiocytes and plasma cells
- usually self-limited, and in cervical lymph nodes
- peripheral blood has atypical lymphocytosis (Downy cells) which are CD8+ T cells
Early: Reactive follicular and monocytoid B-cell (MBC) hyperplasia
- monocytoid B-cell (MBC) hyperplasia seen in early CMV or EBV infection, Toxoplasma, HIV-associated LAD, or non-specific benign hyperplasia
- MBCs are typically negative for BCL2, whereas they are positive in marginal zone lymphoma
- DDx: marginal zone lymphoma -- has more extensive proliferation with confluence and architectural effacement, absence of other reactive patterns of lymphoid hyperplasia, is positive for BCL2 by IHC (benign monocytoid B-cells are negative for BCL2) and is monoclonal (by flow, IHC, or ISH)
Late: prominent paracortical hyperplasia with numerous scattered immunoblasts and hypervascularity
-- DDx: CHL, large cell lymphoma
Micro:
- expanded paracortical area
- sheets and clusters of immunoblasts may mimic DLBCL
-- these immunoblasts are CD30+, CD15-, ALK1 neg, EMA neg, CD45+ and EBV+, and some are T-cells (PAX5+) and some are T-cells (CD3+)
Labs: positive monospot / heterophile antibody, acute infection has serologies consistent with recent infeciton (EBV IgM and IgG)
- RT-PCR shows a high viral load (a positive viral load in the setting of negative serologies = acute infection), although patients with EBV-associated lymphomas will also have a high viral load
- peripheral blood with atypical lymphocytes, which are actually T-cells reacting to infected B-cells and can show an inverted CD4:CD8 ratio by flow cytometry
- remote infx has IgG EBNA and IgG VCA
- gene rearrangement studies: (Oligo)clonal pattern for T-cell receptor may be present
IHC:
Immunoblast phenotype: CD20+ B-cells co-expressing CD30 and MUM1, negative for CD15, EBV-EBER positive (predominantly in interfollicular areas, 10-70% of cells, median 30%) [3]
- absence of BCL6 on the large cells that are positive for MUM1 should be a good clue
- helpful / reassuring features: polyclonal Kappa / Lambda expression by IHC
- retention of OCT2 and BOB.1
- inverted CD4:CD8 ratio in the background of T-cells
DDx: DLBCL - a clue to help distinguish benign infectious mono from DLBCL is that in the benign cases of mono there may be areas with mixed inflammation in the background







Upper left shows scattered reactive follicles, upper right shows fairly well-differentiated mantle zones, lower left shows aggregates of monocytoid B-cells (small cells) in the paracortical region, and the lower right is a somewhat atypical immunoblastic cells (would stain positive for EBV ISH)
High-powered view of monocytoid B-cells (MBCs) in infectious mono, which have slightly irregular, almost centrocyte-like nuclei with abundant pale cytoplasm and some interspersed neutrophils
IM
Features of infectious mono, all cases showed architectural distortion, but showed areas of preserved architecture, which often merged with more abnormal areas
Immunoblast phenotype: CD20+ B-cells co-expressing CD30 and MUM1, negative for CD15, EBV-EBER positive (predominantly in interfollicular areas, 10-70% of cells, median 30%) [3]
- absence of BCL6 on the large cells that are positive for MUM1 should be a good clue
Kappa / lambda is polyclonal by ISH. The large immunoblasts best shown by IHC are also polyclonal for both kappa and lambda.
Infectious mononucleosis with areas of preserved architecture with reactive follicles, patent subcapsular sinuses, and intact capsules. Despite relative hyperplasia of paracortex, always has areas of preserved normal architecture
Infectious mononucleosis. Admixed population of cells of varying sizes. In areas where immunoblasts predominated (right) which could be a florid expansions, like large aggregates or sheets of cells with mitotic activity. Spend some time on low power to appreciate the normal architecture, try to find areas with a variety of cell types. Immunoblasts may resemble Reed-Sternberg (RS) cells (inset). [3]
1 - 8
<
>
Case of infectious mononucleosis of the tonsil with necrosis and ulceration of the epithelium, with a RS-like cell that appears similar to CHL, with CD30+ cells, although the CD30+ cells are of varying size and shape, which is different from CHL, the EBER-ISH highlights the RS-like cells, but there is a greater variability in the shpe of the RS-like cells

Histoplasma Lymphadenitis
-



histoplasma GMS [7]
histoplasma necrotizing granulomas [7]
histoplasma touch prrep [7]
histoplasma see organisms [7]
1 - 4
<
>
Cryptococcus Lymphadenitis
-


Toxoplasma Lympadenitis
Infection with Toxoplasma gondii. One of the MC parasitic dz's of humans; typically in the posterior cervical LNs of young women, which are firm and moderately enlarged
- lymphadenopathy occurs in the late stages of acute infection
- most cases subclinical and do not require tx
Micro: the triad -
1) reactive follicular hyperplasia c lots o mits and phagocytosis of nuclear debris;
2) small granulomas made of pink epithelioid histiocytes in the paracortex that encroach and involve the GCs
- should NOT have well-formed granulomas (which you may see with sarcoidosis), multinucleated giant cells, or necrosis
3) distention of sinuses by monocytoid B cells
- not common to see find T gondii organisms
Dx: should confirm suspicion c Sabin-Feldman dye test, toxoplasma IHC, or IgG immunofluorescence ab test PCR


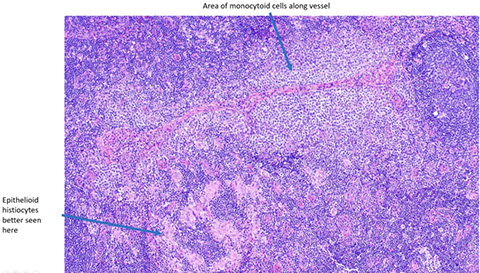

Toxoplasma lymphadenitis. Has lots of reactive follicles, intact capsule with patent subcapsular sinus getting closer to hilum. Increased follicles almost back to back, but have well-preserved and well-defined mantle zones and preservation of the interfollicular areas. There are lots of pinkish cells, which are epithelioid histiocytes which surround and encroach upon involved follicles [3]
Epithelioid histiocytes in toxoplasma infection [3]
Monocytoid B-cells in Toxoplasma lymphadenitis [7]
Classic triad of monocytoid cells, enlarged follicles with reactive germinal centers, and epithelioid histiocytes encroaching germinal centers in Toxoplasma [7]
1 - 4
<
>
Coccidioides Lymphadenitis
-
Herpes Simplex Lymphadenitis
-
Cytomegalovirus (CMV) Lymphadenitis
- Cytomegaly with viral inclusions, which can be seen in moncytoid B-cells (MBCs) in immunocompetent, in paracortex in immunocompromised, and are generally seen in endothelial cells, sometimes T cells or M0 cells

HIV-related lymphadenopathy
May be confused with follicular lymphoma
- with lymphocyte depletion, have dec follicular size
Some say that the follicles here look like continents or animal crackers
3 flavors:
1) Follicular hyperplasia - irregular GC, naked GC, follicle lysis, monocytoid B-cells
2) Follicular involution - regressed GC, thin mantle zone, histiocytes, immunoblasts
3) Lymphocyte depletion - small LN, no follicles, histiocytes, erythrophagocytosis
HIV related lymphadenopathy with follicular hyperplasia showing large irregular germinal centers surrounded by inc clear monocytes
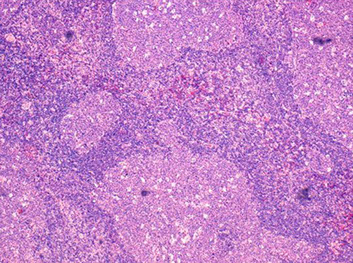
HIV related lymphadenopathy, follicular involution
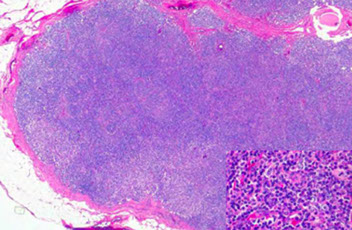
Florid follicular hyperplasia in HIV, looks like animal crackers [7]

Reactive Lymphadenopathies
Inflammatory Pseudotumor of Lymph Node
-

Progressive Transformation of Germinal Centers (PTGC)
B9 reactive condition that causes partial effacement of LN architecture; usually young adults, unknown etiology, M>F
- cervical LN in 50%
- assoc c reactive follicular hyperplasia
- mantle zone lymphs migrate to and disrupt germinal centers (GCs) in multiple areas of the nodules
- no monotypic B-cell population
Micro: large follicles 3-5x the normal size of adjacent reactive follicles, show hyperplastic germinal centers
- interfollicular region typically uninvolved
IHC: GCs CD10+, BCL6+, BCL2-; mantle zones IgD+, BCL2+, CD10-, BCL6-
DDx: NLPHL, LRCHL nodular variant, FL floral variant, HIV-assoc lymphadenopathy, reactive follicular hyperplasia
Px: usually resolves spontaneously
- although related to Nodular Lymphocyte Predominant Hodgkin Lymphoma (NLPHL), no data supports transformation of PTGC to NLPHL
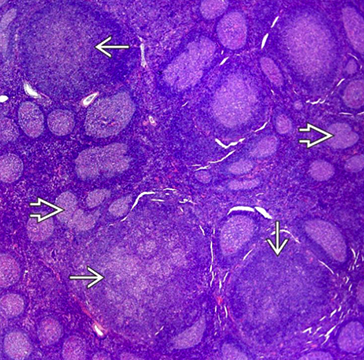


In this case of progressive transformation of germinal centers, there are 3 progressively transformed follicles (solid white arrow) These nodules are 3-4x larger than usual hyperplastic follicles (open white arrows) and are composed of reactive germinal centers infiltrated and disrupted by small lymphocytes.
PTGC, inward expansion of GCs [7]
PTGC IgD [7]
1 - 3
<
>
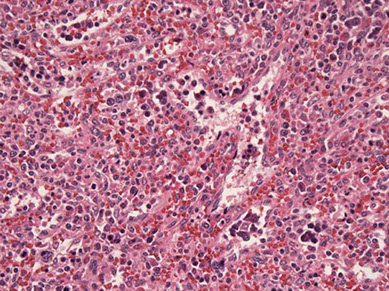
Kikuchi-Fujimoto disease

Kikuchi-Fujimoto disease (KFD)
- aka Histiocytic Necrotizing Lymphadenitis
First described in young women in Japan/Asia, but affects all ages and sexes; presents as persistent painless cervical lymphadenopathy c fever
- usually b9 and self-limited; no known etiology (possibly viral); important to r/o malignancy
- dx can be made on FNA showing plasmacytoid dendritic cells
- lab tests for organisms neg
- can look similar to SLE
3 phases
1) Proliferative - Crescentic histiocytes (+CD68), plasmacytoid denditic cells (+ CD123/303, TCL-1), mostly CD8 T cells, no plasma cells, not simialr to SLE
- reactive follicular hyperplasia (RFH) predominates in early lesions with patchy paracortical expansion
- immunoblasts more apparent (can be sheet-like, as seen in EBV infection), single cell necrosis with some karyorrhectic debris typically present
-- DDx: large cell lymphoma, immunoblasts in Kikuchi can be T-cells, which can raise the possibility of a peripheral T-cell lymphoma (PTCL)
2) Necrotizing - more mature nodules with focal to extensive areas of frank geographic necrosis, homogenous eosinophilic appearance with abundant karyorrhectic debris
- has necrosis c marked apoptosis
- acute inflam absent
3) Xanthomatous - resolution phase with numerous foamy macrophages
Micro: - nodules contain variable mixture of immunoblasts and histiocytes and plasmacytoid dendritic cells; few plasma cells, neutrophils consicuously absent
- lots of necrotic debris, scattered fibrin deposits, and mononuclear cells trying to clean it up
Plasmacytoid dentritic cells and activated T cells are numerous (though plasma cells and neutros are few)
- useful feature is the lack of neutrophils, plasma cells, and eos
Plasmacytoid dendritic cells are IFNa-producing cells with role in antiviral immunity
- resemble plasma cells, dispersed (not condensed) nuclear, chromatin
- Frequently observed in KFD (cluster at edge of nodules), viral infections, Castleman's disease
IHC: highlights predominance of CD3 cells and macrophages
- Plasmacytoid dendritic cells -- medium-sized with monotonous, round to oval nuclei, and basophilic cytoplasm, CD2+, CD4+, CD43+, CD123+, CD68+ (weak), CD3-
- Immunoblasts -- large with vesicular round nuclei and prominent nucleoli - CD3+, CD8+ > CD4+, many TIA+, granzyme+
- Histiocytes -- medium-sized with oval or crescent-shaped nuclei, foamy of debris-laden cytoplasm, are CD68+ (strong), MPO+, lysozyme+
Px: excellent -- resolves spontaneously
DDx: Peripheral T-cell Lymphoma NOS -- PTCL has completely effaced architecture, monomorphous, lack of characteristic admixture of cell types including crescentic / foamy histiocytes and PDCs
- Nodal PTCL-NOS usually CD4+ vs CD8+ immunoblasts in KFD
- Beware of diagnosing PTCL-NOS in a child or young adult --> exceedingly rare in this age group!!!



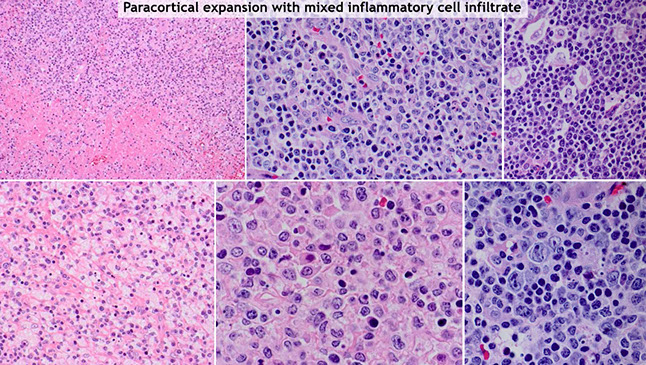







Kikuchi-Fujimoto disease [3] - Classic low-power appearance with reactive follicles at the periphery and paracortical expansion.
Kikuchi-Fujimoto disease [3]
Maturation of necrotizing process in KFD
Kikuchi-Fujimoto disease [3] - histiocytes can have a crescent forms (bottom middle), or xanthomatous forms (bottom left) in later (cleanup) phases, immunoblasts with prominent nucleoli and vesicular chromatin (bottom right), and plasmacytoid dendritic cells (top middle) which are CD123+, and necrosis (top left)
May look like reactive follicles, but are not reactive follicles because do not have well-formed mantle zones
KFD with characteristic necrosis, crescentic histiocytes, clusters of PDCs, and MPO staining in histiocytes
KFD. T immunoblasts with strong CD3+, with CD8 predominance over CD4, though CD4 positive in T-cells and histiocytes in background. PAX5 is positive in residual follicles, less so in paracortical areas [3]
KFD histiocytes - CD68 strongly +, MPO with cytoplasmic staining
KFD characteristics
KFD IHC
KFD. "Maturation" of necrotizing process
1 - 11
<
>
MBCs tend to be more irregular in shape, have mature mature rather than dispersed chromatin, somewhat more abundant cytoplasm, and have a sprinkling of neutrophils

Rosai-Dorfman disease (RDD)
- aka sinus histiocytosis with massive bilateral lymphadenopathy (SHML)
Massive painless bilateral LN enlargment in neck of teenager c fever, leukocytosis, inc ESR, and polyclonal inc IgG usually in black kids/teens (though can occur at any age)
- traditionally considered reactive, non-neoplastic proliferation of unusual subset of macrophages
- etiology unknown, but infectious origin suggested
Rare; children/young adults (mean 20 yrs).
•More common in males of African descent.
•Bilateral painless massive cervical LAD; fever, weight loss.
•mediastinal, inguinal, retroperitoneal LN may also be involvement.
•Extranodal involvement in ~40% of cases [skin, nasal cavity, soft tissue, orbit, bone (lytic lesions), salivary gland, CNS, breast, pancreas].
- anemia with lymphopenia may be seen at diagnosis
- has characteristic cytologic features with striking emperipolesis
__________________________________________
Gross: nodes bound together by fibrosis c gray to yellow cut surface
__________________________________________
Micro: dilated sinuses c lymphs, plasma cells, and foamy histiocytoid cells c vesicular nuclei and lots of pale cytoplasm
- histiocytes have emperipolesis / lymphocytophagocytosis (they ate some lymphs)
- effacement of architecture by dilated sinuses
- capsular / pericapsular fibrosis and inflam is common
Lymph node: fibrotic capsule, massive dilatation of sinuses filled with characteristic histiocytosis with large nuclei and prominent nucleoli.
•Histiocytes: large with abundant pale cytoplasm showing emperipolesis.
•Positive: S100, histiocytic markers (CD68, CD163, CD4, CD11c, CD33, fascin), Cyclin D1 (often aberrantly expressed in the nuclei of clonal / neoplastic histiocytic proliferations), OCT2
•Negative: CD1a, CD207 (Langerin), BRAF V600E.
•Numerous plasma cells in the background (polytypic); helpful feature in extranodal sites.
•Frequently contain numerous IgG4 positive plasma cells; DDx IgG4-related disease.
__________________________________________
Genes: Negative for clonal IgH or TCR gene rearrangements.
•Polyclonal by HUMARA assay.
•Lack BRAF V600E mutations.
•Rare cases with NRAS/KRAS mutations recently reported.
__________________________________________
IHC: Oil red O shows lots of neutral lipid in the histiocytes, which are also strongly (+) for S-100 and CD68, PLAP+, but negative for CD1a and EBV
- OCT2 is a sensitive (+ in 97%) marker for abnormal histiocytes of Rosai-Dorfman disease
-- oct2 is + in 7% of LCH and 0% of ECD
__________________________________________
EM: histiocytes in the sinuses have extensive pseudopodia and lack Birbeck granules
__________________________________________
DDx:
- Reactive lymph node with sinus histiocytosis
- Hemophagocytosis
- Langerhans cell histiocytosis
__________________________________________
Tx: usually has fast and spontaneous relapse
- chemo may be helpful
__________________________________________
Px: Generally regarded as a non-neoplastic, reactive process with self-limited/indolent disease course, especially in classical (nodal) form.
•5-10% of pts may die of disease due to organ involvement or immunologic abnormalities.
Rosai-Dorfmann dz





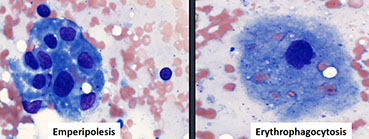


* more prominent in extranodal sites


Touch prep from patient with Rosai Dorfman, histiocyte with abundant eosinophilic cytoplasm with ingested matieral. Cell was CD14+, CD68+, S100+, and CD1a- [6]

Rosai-Dorfman disease [6]. Looks vaguely whorled from low power


Rosai-Dorfman disease, S100 staining nucleus and cytoplasm, brings out emperipolesis [6]


Kimura disease
Endemic in Asia; presents as mass lesion in head / neck or major salivary glands usually in males
- has features that overlap c Angiolymphoid hyperplasia with eosinophilia (ALHE); except AHLE is mostly in the skin of white girls (no lymphadenopathy)
Micro: LNs have marked germinal center hyperplasia (some of which are progressively transformed)
- germinal centers have lots of BVs, interstitial fibrosis, and proteinaceous material, sometimes c eosinophilic abscesses
- see some paracortical sclerosis c hyalinized vessels
Kimura disease with polykaryotes

Castleman disease
aka giant LN hyperplasia
- similar labs as angioimmunoblastic lymphadenitis
May be solitary or multicentric
-- solitary MC presents in mediastinum and is asymptomatic (though can have sx if plasma cell type)
-- multicentric is nearly always plasma cell type and has generalized lymphadenopathy c spleen
- multiple germinal centers in 1 follicle (aka twinning or atretic GC)
- lollipop sign - radial penetrating arterioles
- onion skinning: concentric layers of mantle zone lympocytes
Nonlymphoid cells are involved in pathogenesis, such as follicular dendritic cells; and follicular dendritic cell tumors may develop
2 flavors: hyaline-vascular type or plasma cell type
1) Hyaline-vascular / solitary / unicentric type
Large follicles c lots of BVs and hyalinization or plasma cells in paracortex in mass of lymph tissue that may look like a Hassal corpuscle
- has vesicular cells in hyalinized area that are follicular dendritic cells (+ CD21/23/35)
- surrounded by concentric layers of lymphs (like a mantle zone) which looks like onion skin
- sinuses are absent
- lymphoid sub-subtype has larger mantle zone and smaller germinal center
- no assoc c HHV-8
- tx is surgery
- Px good
2) Plasma cell / diffuse / Multicentric type
- aka Multicentric castleman dz
Lots of plasma cells in interfollicular tissue, possibly c Russell bodies
- has amorphous acidophilic material (probably fibrin) and no hyaline-vascular changes
- assoc c HHV8 (sometimes seen c Kaposi sarcoma) which causes inc serum IL-6 (HHV-8 encodes IL-6), POEMS syndrome, HIV, Wiscott-Aldrich syndrome, myasthenia gravis, pemphigus
- inc freq of plasmablastic lymphoma, Kaposi sarcoma, primary effusion lymphoma
Tx: chemotherapy
Px: Poor
Interfollicular plasmacytoid monocytes (+) CD123

Castleman dz, lollipop sign


Castleman dz, hyaline vascular variant with "twinning" with 2 or more atrophic GCs (white arrows)
HHV8+ multicentric Castleman disease. A. B-cell follicle with a regressed, partially hyalinized germinal center. B. Mantle zone has scattered plasmablasts. C. B-cell fiollicle with partial hyalinization of the germinal center with an attenuated mantle zone. D. Germinal center with numerous plasmablasts
Rheumatoid Arthritis-Related Lymphadenopathy
-
Sarcoid Lymphadenopathy
-


Dermatopathic Lymphadenitis (DL)
- aka lipomelanosis reticularis of Pautrier, or dermatopathic lymphadenopathy
Nodular paracortical hyperplasia secondary to generalized dermatitis c pruritis, usually c exfoliation (rarely occurs without assoc skin dz or rash)
Micro: paracortical aggregates of histiocytic / dendritic cells, including Langerhans cells and melanin-containing macrophages
- inc T cells in cortex / paracortex, Langerhans (LC) / interdigitating dendritic cells (IDC) c melanin-laden macrophages
- interdigitating dendritic cells c elongated, twisted nuclei c nuclear grooves, pale chromatin, and lots of eosinophilic cytoplasm
- post-capillary venules may be prominent
- has pale nodular paracortical areas
- see peripheral blood eosinophilia
IHC: (+) S100, Fascin (IDCs)
- CD1a and Langerin highlights the Langerhans cells
- Fontana-Masson can highlight melanin pigment
DDx: Mycosis fungoides (T cell ag loss, TRB or TRG rearranged, and vascular prolif)
- Langerhans cell histiocytosis (LCH) when florid -- LCH involving lymph nodes shows intrasinusoidal (not solely paracortical) distribution with sinusoidal distention; to help differentiate, Cyclin D1 (and p-ERK) are expressed by neoplasatic cells of LCH, but not significantly by reactive Langerhans cell proliferations, and BRAF V600E is positive in ~50% of LCH



Dermatopathic lymphadenitis
Dermatopathic lymphadenitis, patient with history of nearby skin lesion that drains to this lymph node [7]
Differential diagnosis: Dermatopathic Lymphadenitis (DL) versus Langerhans Cell Histiocytosis (LCH)
In LCH the collection of Langerhans cells are restricted mostly to the sinuses, which may not be easily appreciated on H&E, there may be spillover into the paracortical area (seen with the langerin stain), the cyclin D1 stain shows how the cells are mostly sinusoidal
2 - 3
<
>
Hemophagocytic lymphohistiocytosis (HLH)
-aka macrophage activation syndrome or hemophagocytic syndrome [9]
Caused by systemic activation of macrophages and CD8 Tc cells, and assoc c T-cell lymphomas esp subcutaneous panniculitis like T-cell lymphoma
- macrophages eat up progenitor cells in BM and other stuff in peripheral tissues and release inflammatory mediators (TNFa, IL-6/12) that causes systemic inflammation leading to cytopenias and sx that look like shock
- may be genetic, in which case Tc and NK cells are not made properly and cytotoxic granules are released willy-nilly into circulation
- can also be caused by viruses, T cell lymphoma, x-linked syndrome
__________________________________________
Criteria (need 5 of 8):
Clinical: Fever, hepatosplenomegaly
Hematologic: Cytopenias (>=2 of 3), Hemophagocytosis
Serum values: Hypertriglyceridemia, Hypofibrinogenemia, Hyperferritinemia
Immunologic: Low or absent NK function, Elevated soluble CD25
Additional: Molecular demonstration of known gene mutation, Presence of a familial disease
__________________________________________
Severely ill patients, any age
- clinically there is a differential with an acute leukemia
- fever hepatosplenomegaly, LFT abnormalities, adenopathy, other constitutional sx
Disseminated macrophage proliferation and activation
- biologically it is an abnormal function of NK cells and T-cells
- macrophage proliferation and activation with exuberant cytokine release ("cytokine storm")
May be primary (inherited) or secondary
Familial Lymphohisitocytosis (primary lymphohistiocytosis)
- presents in infancy or early childhood
- mutations in several genes known (perforin, MUNC13-4, Syntaxin)
- similar syndromes occur in other immunodeficiencies (XLP, Chediak-Higashi, Griscelli syndrome)
Secondary - essentially all adult hemophagocytic syndromes are secondary
- infection associated - viral (EBV), bacterial fungal protozoal
- malignancy-associated (paraneoplastic from Hodgkin lymphomas)
= autoimmune-associated -- esp juvenile rheumatoid arthitis, "macrophage activation syndrome"
__________________________________________
Micro: bone marrow biopsies show histiocytic hyperplasia with marked hemophagocytosis
- findings are similar regardless of underlying cause
- look closely for underlying lymphoma in adults
__________________________________________
DDx: neoplastic hisitocytic disorders
- large cell lymphoma
- reactive increase in BM histiocytes / monocytes (increased cell turnover, MDS, MPN, granulomas, histiocytic hyperplasia associated with lymphoma, fat necrosis (post-chemotherapy), growth factor therapy)
__________________________________________
Px: Fulminant clinical course, poor without treatment
__________________________________________
Tx: immunosuppression and cytotoxic agents, sometimes may need bone marrow transplant
- without therapy, high mortality rate
Hemophagocytic lymphohistiocytosis (HLH)
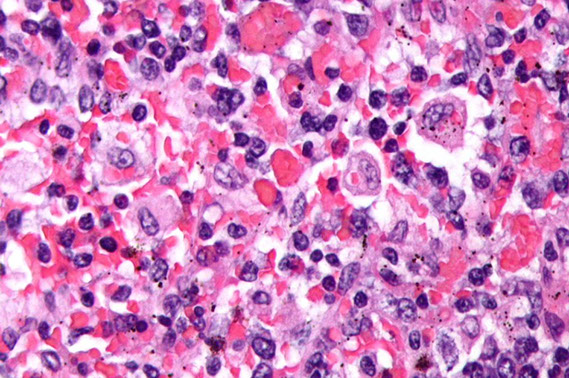
Clinical and Laboratory criteria for the diagnosis of HLH [9]

Florid HLH [13]. Bone marrow aspirate with markedly increased histiocytes with prominent hemophagocytosis with ingestion of RBCs and neutrophils (usually it is not thils florid, will probably only see 1-2 histiocytes eating blood)

Lymphadenopathy Associated with Joint Protheses
-
Lipid-Associated Lymphadenopathy
-
Lymphadenopathy Secondary to Drug-Induced Hypersensitivity Syndrome
-
IgG4-Related Disease
-
Immunodeficiency-Associated Lymphoproliferations
Overview of Immunodeficiency-Associated Lymphoproliferative Disorders
-
ALPS

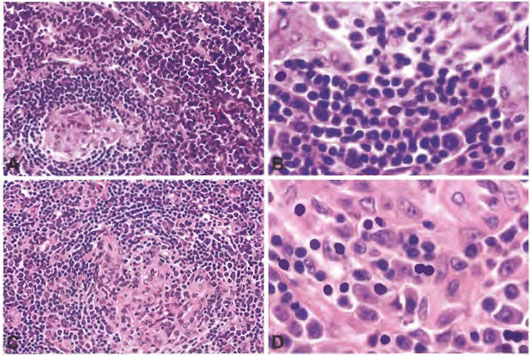
Autoimmune lymphoproliferative syndrome (ALPS)
Germline (sometimes somatic) mutations of FAS (TNFRSF6), FASL, or CASPASE gene that causes splenomegaly, hyperIgG, and autoimmune sx mostly in kiddos
- inc CD3+CD4-CD8- (double negative) alpha/beta T cells
Micro: paracortical expansion by smaller alpha-beta double negative T-cells c larger immunoblasts that can mimic lymphoma
- cells are CD45RO negative, which makes them cirgin T-cells
- florid follicular hyperplasia
- can have RDD-like changes
lymphoid hyperplasia in T-cell areas of spleen

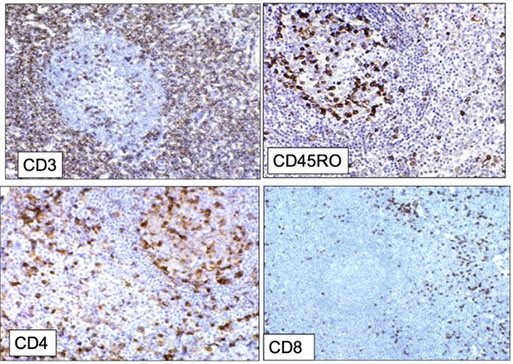
ALPS IHC: paracortical T-cells are negative for CD45RO, CD4 and CD8, looks like some aberrent expression, but it is NOT! Be very careful of diagnosing PTCL in young patients!
ALPS - pt with marked splenomegaly, protuberant abdomen and marked cervical LAD , biopsy shows some reactive follicles with abundant paracortical expansion [4]
Immunomodulating Agent-Associated Lymphoproliferative Disorder
-
Posttransplant Lymphoproliferative Disorder, Early Lesions and Polymorphic
-
Posttransplant Lymphoproliferative Disorder, Monomorphic
-
Nonhematopoietic Proliferations in Lymph Node
Epithelial Inclusions in Lymph Node
Nevus Cell Inclusions in Lymph Node
Vascular Transformation of Lymph Node Sinuses
Angiomyamatous Hamartoma
Palisaded Myofibroblastoma
Metastatic Kaposi Sarcoma
Epithelial Inclusions in Lymph Node
-
Nevus Cell Inclusions in Lymph Node
-
Vascular Transformation of Lymph Node Sinuses
-
Angiomyamatous Hamartoma
-
Palisaded Myofibroblastoma
-
Metastatic Kaposi Sarcoma
-
Spleen
Note: Primary lymphoid organs = BM and thymus
- site of lymphocyte birth and maturation by a process that does not involve antigens; have no follicles and make mature naive lymphocytes
Secondary lymphoid organ (LNs, spleen, MALT) activate naive lymphocytes with antigen and have follicles
Embryology
Found @ ~5 WGA, red and white pulp distinguished at 9 mo GA
- functions as site of development of blood cells until ~6 mo GA (but hematopoiesis does not occur in spleen, just development)
- accessory spleens seen in ~ 1/4 of autopsies
- immotile cilia syndrome assoc c polyspenia
- lots of apoptosis in germinal centers of B-cell follicle, and is also T cell maturation
Anatomy / Physiology
Covered by a capsule and peritoneum, c dense irreg connective ittsue (has smooth muscle in some animals which can contract and give blood in emergencies)
- Splenic artery (from celiac artery) goes into hilum and gives rise to BVs from capsule (trabecular arteries and veins) supply blood to red pulp
- arteries leaving the capsule engulfed by periarteriolar lymphoid sheath (PALS), which goes into a white pulp nodule where it is then called the central artery (follicular arteriole)
- when central artery leaves the white pulp becomes the penicillar artery, which end as capillaries ensheathed by macrophages
- 90% of RBC removal occurs in the spleen. Cells removed include:
Senescent cells
Cells with decreased deformability
Opsonized cells
Cells with inclusions that cannot be removed by grooming
Cells with intracellular parasites (malaria, babesia, etc.)
Cells with crystals (e.g. Hb C) or increased viscosity (sickled cells)
Severely damaged or misshapen RBCs are removed by the liver
The function of littoral cells is to maintain a healthy erythron.
They do this in three ways: grooming, pitting, and culling.
Grooming is the removal from the cell’s surface of excess:
lipids ; cholesterol ; and proteins
Grooming prevents the formation of target cells, stomatocytes, acanthocytes
Pitting is removal of intracellular particles from RBC while leaving the cell intact.
Inclusions removed include:
Howell-Jolly bodies ; Heinz bodies ; Pappenheimer bodies ; intracellular parasites
“Bite cells” may be the result of pitting
Culling is removal of damaged or senescent RBCs, known as erythrophagocytosis
- Aging RBCs undergo morphologic changes that signal senescence.
- Protein Band 3 clusters in cell membrane, appears to be “non-self”
- RBCs are opsonized
Littoral cells phagocytize the opsonized cells.
There are two major factors responsible for age related changes in RBCs that will initiate phagocytosis in the spleen:
Decrease in enzyme activity
ATP becomes less available as EM pathway shuts down
Na + /K + pump less efficient --> More internal Na+; Less internal K +
Greater Internal viscosity
- Phosphogluconate pathway shuts down --> Less G6PD activity causing more reactive O2 species, more unstable hemoglobin
- Luebering-Rapaport Pathway shuts down --> Less 2, 3 DPG made, Oxygen affinity increases
Repeated passes through the spleen
reduces membrane phospholipids - Cholesterol content reduced ; Membrane becomes rigid
- Parts of the cell membrane are removed: MCHC increases ; MCV decreases (younger cells are larger) ; Cells become more spheroidal
- Decreased sialic acid --> Cells become more agglutinable
Spleen also serves as a reservoir for platelets
- 30% of circulating platelets are sequestered in the spleen
-- allows immediate release to the circulation if needed
Splenomegaly (increase in the size of the spleen) can cause increased sequestration of platelets
Causes of splenomegaly: Cirrhosis of the liver; Leukemia or lymphoma
Splenectomy
can occur two ways: Surgery ; Auto-splenectomy
Patients with no spleen have an increased risk of infections, particularly from encapsulated bacteria.
- Peripheral blood findings in splenectomized patients:
Codocytes (Target Cells) ; Howell-Jolly bodies ; Acanthocytes ; Stomatocytes ; Spherocytes
Indications for Splenectomy
•Trauma/Incidental
•Myeloproliferative Neoplasms
•Lymphoproliferative Disorders
•Staging Laparotomy
•Hereditary Spherocytosis
•Splenic Lipidoses
•Autoimmune Disorders
•Diagnosis of Splenic Mass (CT, MRI)
•“Hypersplenism”
Histology
Lacks cortex and medulla
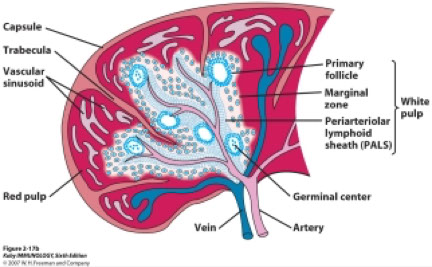
The white pulp is involved with immune surveillance.
It is composed of nodules called Malpighian corpuscles
These corpuscles are composed of "lymphoid nodules" (or "follicles") which are rich in B-lymphocytes, and periarteriolar lymphoid sheaths (PALS) which are rich in T lymphocytes
- irrigated by central artery (surrounded by PALS) which gives off radial arterioles, that end in marginal sinus (goes around the white pulp)
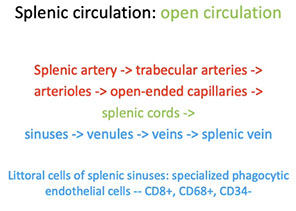
- marginal sinus and central artery blood go into penicillar arteries ending in capillaries surrounded by macrophages (macrophage-sheathed capillaries), which can drain into splenic sinusoids (closed circulation) or red pulp stroma (open circulation)
The red pulp is where the red blood cells are filtered.
It consists of long, tortuous sinuses which the red blood cells must filter through
Red pulp = penicillar arterioles, macrophage-sheathed capillaries, (splenic) cords of Billroth [reticular cells forming stroma], and splenic (venous) sinusoids, and all types of blood cells stored here
- cords of Billroth lined by macrophages, plasma cells and RBCs(?), separates sinusoids
- sinusoids surrounded by macrophages that filter old RBCs and circulating bugs from the blood (similar to Kupffer cells in liver)
-- sinusoids surrounded by ringed-network of reticular fibers and basal lamina
Post-splenectomy, may see Howell-Jolly bodies (nuclear remnants), target cells, and thrombocytosis
***T cells found in structures that begin with "P" = PALS in spleen and Paracortex of LNs; B cells found in follicles - follicles of cortex in LNs and follicles of white pulp in spleen***
Gross
Weigh / measure, assess hilar vasculature, LNs
- photograph capsular abnormalities
- section at 1 cm, photograph abnormalities
- sample splenic parenchyma (1-2 sections if normal, more if abnormal)
- submit hilar LNs
- Sample gross lesions (lacerations, cyst wall, mass lesions)
- if lymphoma suspected, do touch prep and flow cytometry / cytogenetics


Splenic circulation



Normal spleen. Slice showing intact capsule, no thrombi in vessels, red pulp with small nodules of white pulp






Splenogonadal fusion typically occurs with the left testis

Spleen capsule [5] - is the only thing that stands between the entire blood volume or your spleen and the abdominal cavity


Diffuse Splenic Lesions
White Pulp - Miliary Pattern
•Reactive lymphoid hyperplasia.
•Granulomas.
•Small B-cell lymphomas (e.g. MZL, CLL, FL).
Diffuse WP miliary pattern
•Splenic marginal zone lymphoma.
•Micronudular variant of T-cell histiocyte-rich large B-cell lymphoma.
Diffuse RP pattern
•Hairy cell leukemia.
•Splenic diffuse red pulp small B-cell lymphoma.
•Hairy cell leukemia variant.
•Hepatosplenic T-cell lymphoma.
Red Pulp - Diffuse "beefy red"
•Storage disorders.
•Extramedullary hematopoiesis.
•Myeloid and lymphoid leukemias (e.g. AML, MPN, hairy cell leukemia).
•Histiocytic proliferations
Focal/multifocal Splenic Lesions
Large cell lymphoma
Hodgkin lymphoma
Pseudotumoral lesions (IPT, hamartoma, SANT)
Vascular lesions/tumors
Metastatic tumors (carcinoma, sarcoma)
Reactive (cysts, peliosis, infarcts)
Extramedullary hematopoiesis
When the bone marrow is compromised or overwhelmed and cannot produce enough red blood cells to support the erythron, the spleen will take over erythropoiesis.
Causes: bone marrow infiltration from myelofibrosis or leukemia
- Clinical signs of a compromised bone marrow: Nucleated RBCs ; Teardrop cells
Splenic Injuries
Mechanical
Trauma - laceration, hemorrhage, "fractured spleen"
- implants can grow into splenic tissue ("splenosis")
Infarction - embolic, sickle cell disease, torsion
Congestion - Acute, chronic (with fibrosis -- cirrhosis, portal HTN)
Atraumatic splenic rupture
- Neoplastic - 30% -- homorrhagic / vsscular
- Infectious 27% -- EBV, malaria
- inflammatory 20% -- pancreatitis, vasculitis
- drug - tx 9% -- GCSF included
- Mechanical 7% -- pregnancy, cirrhosis
- Normal spleen 6%

Splenic infarct with necrosis with surrounding organization

Splenosis - small splenules with a capsule with prominent EMH

Splenic fracture from MVA

Autoimmune Splenitis
Splenic capsule is thick, known as hyaline perisplenitis (or sugar-icing)
- mainly a white pulp problem, centered around blood vessels, with white pulp hyperplasia and eventually scarred blood vessels
- the red pulp is fairly normal in autoimmune disorders involving the spleen
DDx: may have some cellular breakdown that looks a little like Kikuchi-Fujimoto dz
Autoimmune splenitis - forms nodule around vessel that replaces the white pulp. First shows onion-skinning, then these scar-like vessels

Splenic Cysts
Secondary Cysts (eg post-trauma
- lack lining, thin-walled, don't recur
Primary cysts
- thick fibrous wall
- squamous, mesothelial, or rarely transitional epithelium (may be denuded)
- recurrence risk if incompletely removed
- can cause elevation in CA19-9, CEA
Parasitic cysts (echinococcal) - contain scolices

Primary splenic cyst

Reactive abnormalities of the red and white pulp: consumption [5]
Autoimmune hemolytic anemia
Immune thrombocytopenic purpura
- splenectomy usually follows exhaustion of pharmacologic options
Hemophagocytic lymphohistiocytosis (HLH)
- with or without malignancy (often lymphoma)
- splenectomy often in search of lymphoma dx









AIHA post rituximab
AIHA Red pulp
AIHA with congestion and EMH
AIHA, high power of previous. NRBCs floating in the dilated sinuses, lined by littoral cells. In the cords there are numerous RBCs back to back that are trapped
ITP expansion of red and white pulp. Germinal centers are not features of a normal adult spleen
ITP expansion of red and white pulp. Germinal centers are not features of a normal adult spleen. Germinal center surrounded by mantle zone
ITP histiocytosis.Platelet trapping in splenic cords, with histiocytes with fluffy pink cytoplasm
Evans syndrome with florid follicular hyperplasia
The packs of RBCs here is actually within macrophages, which have eaten all the RBCs
9 - 9
<
>
Splenic Vascular Proliferations

Non-hematolymphoid/pseudotumoral lesions presenting as splenic mass:
Splenic cysts
Peliosis
Inflammatory pseudotumor
Splenic hamartoma
Vascular tumors (cord capillary hemangioma, littoral cell angioma)
Sclerosing angiomatoid nodular transformation
Metastatic tumors
Storage disorders
Splenic Hamartoma
•Tumor-like proliferation composed of structurally disorganized mature red pulp elements (cordal histiocytes, vasculature, myoid cells).
•Uncertain etiology.
•Rare (0.2%); usually incidental finding.
•M=F; diagnosed at all ages (11 mo to 86 yr).
•Majority pts asymptomatic.
•Splenectomy curative.
- IHC: CD31+, CD68+, CD8+
Cord Capillary Hemangioma
•Rare tumor with clinical and morphologic overlap with hamartoma.
•Initially considered a morphologic variant of hamartoma (“capillary-rich” variant).
•Different vascular composition from hamartoma (CD8-/CD34+).
•Demonstrated to be clonal and different from hamartoma.
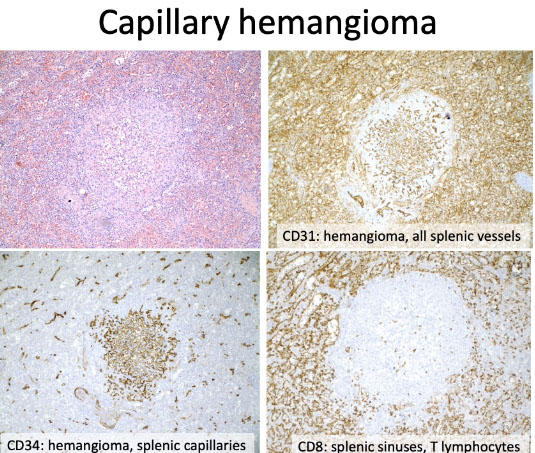



Just a normal Capillary hemangioma, CD31+, CD34+
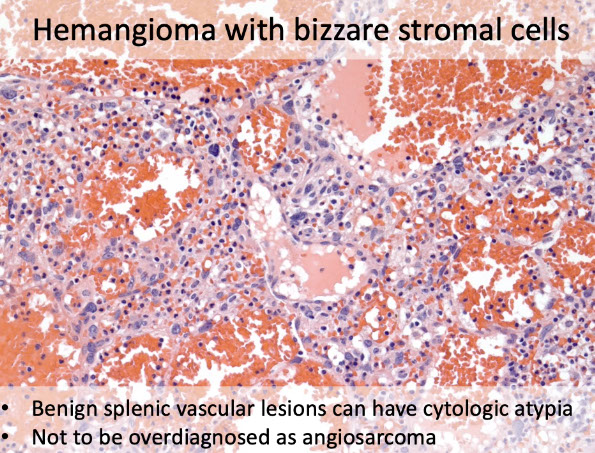
Bizarre capillary hemangioma, don't overcall it!! It does not have more malignant potential [5]
Splenic hamartome, gross.
Splenic hamartome, gross. 2
Splenic hamartome, gross. 3
2 - 5
<
>
Splenic Hamartoma
- has same IHC pattern as normal sinuses


Border between hamartoma and normal spleen disappears at high power because it is basically the same vasculature
Splenic hamartoma. High power. [5] Blends with adjoining red pulp at high power. CD31+, CD68+, CD8+, CD34- sinuses
1 - 2
<
>
Sclerosing Angiomatoid
Nodular Transformation (SANT)
•Non-neoplastic vascular lesion with angiomatoid nodules associated with fibrosis
•May represent reaction to inflammation, tumor, and hemorrhage
•Solitary well-demarcated mass with central scar
•Small vessels with variable phenotype: sinusoidal (CD8+/CD31+/CD34-); capillary-like (CD8-/CD31+/CD34+); venous (CD8-/CD31+/CD34-).
•Some cases show increased IgG4 plasma cells (not considered part of spectrum of IgG4-related diseases)
SANT



SANT [5]. Grossly it can look pretty scary.

Littoral Cell Angioma
•Benign vascular neoplasm of red pulp sinus-lining cells with histiocytic and endothelial cell differentiation.
•Rare; usually incidental.
•Multiple spongy lesions throughout the spleen.
•M=F; all age groups.
•Splenectomy curative.
•Frequent association with visceral malignancies. – Peckova K et al. Histopathology 2016;762-774.
IHC: CD31+/CD68+, unlike normal sinuses, are CD8-, CD21+/-, CD34-






Gross
Littoral cells bulge into vascular lumina
Littoral cell angioma IHC
Mauris sit amet tortor.4
Mauris sit amet tortor.5
Mauris sit amet tortor.6
3 - 6
<
>
Splenic lymphangioma
Should stain with normal lymph vessel markers (CD31+, D2-40+)
Splenic lymphangioma [5]

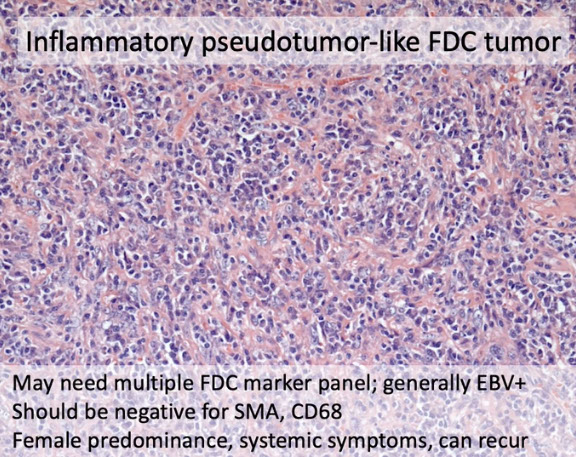
Splenic inflammatory pseudotumor-like FDC tumor
Its name is inversely proportional to its incidence
- May need multiple FDC marker panel,
- generally EBV+,
= negative for SMA and CD68. Female predominance, systemic symptoms, can recur
EBV positivity seen in the stromal cells
Splenic inflammatory pseudotumor-like FDC tumor. May need multiple FDC marker panel, generall EBV+, should be negative for SMA and CD68. Female predominance, systemic symptoms, can recur


General rule: lymphomas that involve the LNs usually involve the white pulp of spleen, and those that involve the blood and BM involve the red pulp [5]
Splenic Marginal Zone Lymphoma (SMZL)
•First described by Schmid et al in 1992
(Am J Surg Pathol 1992;16:455-466).
•<2% of lymphoid malignancies.
•Middle aged to elderly pts without gender predominance.
•Associated with HCV (S. Europe), cryoglobulinemia type II, autoimmune disorders.
Splenomegaly without lymphoadenopathy (although sometimes the spleen can be of normal size!!), lymphocytosis (+/- villous lymphocytes), cytopenias
•BM with intrasinus involvement
- blood commonly involved, see villous lymphocytes
•Peripheral LAD uncommon
•Autoimmune phenomena (20%)
•Monoclonal paraprotein, <20g (33%)
•Median survival 10 years
•May transform to LBCL
Micro: biphasic pattern with prominent marginal zone in spleen (mimics the splenic white pulp)
- medium-sized cells with pale cytoplasm
By definition, the lymph nodes (LNs) involved by SMZL do not show the prominent zonation that is seen in the spleen
IHC:
Positive:
•Pan-B-cell antigens (CD19, CD20, CD22, BSAP/PAX-5), BCL-2, CD24, CD27, IgM, IgD
Variable:
•CD5 (20%), CD23 (30%); CD5/23 co-expression rare
CD5 expressed in up to 20%
- cases of SMZL that are CD5+ are similar to the classic CD5- variant except tend to have higher lymphocyte cound at dx and more diffuse BM infiltration
•DBA.44 (75%), CD11c (50%), CD25 (25%)
•CD138, kappa/lambda (PC differentiation)
•IRTA1 (25-50%), MNDA (70%)
Negative
•CD10, BCL-6, CD43 (important negative finding), cyclin D1, SOX11, LEF1, CD103, annexin A1
Genes: cytogenetic / genotype studies are not usually necessary for the diagnosis, though should have clonal Ig gene rearrangement
Allelic loss of 7q31-32 (30%).
- clonal IgH R, IgH mutations
•Small number of cases show t(2;7)(p12;q21) CDK6-IgK; dysregulation of CDK6 gene at 7q21.
•Trisomy 3q (10-20%).
•17p deletion; p53 allelic loss.
•NO evidence of MALT lymphoma-associated translocations t(11;18)(q21;q21), t(14;18)(q32;q21), or t(3;14)(p13;q32).
A subset may have MYD88 mutations, so cannot use this to differentiate from LPL
Molecular Genetics
•NOTCH2 mutations (10-25%); may also be seen in small number of cases of MALT lymphoma and DLBCL.
•KLF2 mutations (10-40%); also found in other small B-cell lymphomas. (people aren't looking for this now)
•MYD88 mutations rare in SMZL; helpful to distinguish from LPL in cases with plasmacytic differentiation.
•NOTCH2, KLF2, and TP53 mutations reported to be adverse prognostic indicators based on limited data.
DDx:
Major diagnostic pitfalls and how to deal with them
- Hyperplastic splenic marginal zones -- nonclonal , here, genetic studies are critical!!! (FCIPS/genotypic studies), splenic marginal zones can be very prominent in many types of benign spleens
- Pale appearing marginal zone involvement by other types of NHL -- FL, MCL, other MZL
--- morphology: homogeneous centrocytes in FL
- Phenotype: SMZL typically are CD5-, CD10-, CD23-, CD43-, CyclinD1-, although cases studies by FCIPS are more often reported to be CD5 or CD10+
---- IgD positivity is typical but not a specific finding and not always found
Tx: splenectomy may be the treatment of choice even if stage IV
- regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection with interfreon (alone or in combination with ribavirin)
-- Dx not based on splenectomt
- Similar treatment failed in 6 patients with non-HCV associated SLVL
A recent study [Blood. 2016;128(21):2527-2532] has shown that direct-acting antiviral agents are able to induce lymphoma response in patients with HCV-associated indolent non-Hodgkin lymphoma
- the highest rate of lymphoma response (73%) was seen in pts with marginal zone lymphoma
SMZL


Histologically, SMZL mimics the white pulp

SMZL villous lymphocyte
SMZL involving a LN, showing cells with pale cytoplasm, zonation with not be present in the LN as it is in the spleen





A classic case of SMZL, with increased and enlarged white pulp nodules, with dark central cores and paler peripheral marginal zones, also infiltration of the red pulp with little nodules and cells in the cores.
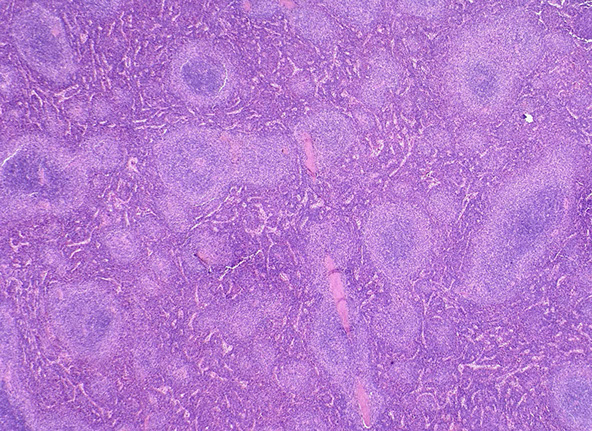
A classic case of SMZL, at higher mag, see the darker central core with lighter marginal zone, with a larger number of smaller lymphs with less cytoplasm in the central cords, here there are too many lymphocytes in the red pulp to be considered normal
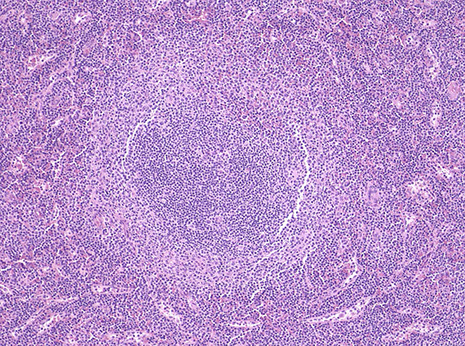
Classic SMZL, high mag showing the small lymphocytes of the central core in the white pulp nodule, and the cells in the marginal zone with more abundant pale cytoplasm that looks like normal splenic marginal zone cells and smaller lymphocytes in the red pulp

Some cases of SMZL have mostly or exclusively monophasic WP nodules, instead of biphasic WP nodules


Splenic marginal zone lymphoma, involves the periphery of the white pulp nodules. paler region where cells are larger, have more cytoplasm and more dispersed chromatin
Marginal zone lymphoma in a mesenteric lymph node Left - follicles iwth GC, smaller mantle zone and paler peripheral zone, high mag (right) shows difficult to tell where mantle zone ends and marginal zone begins, bottom shows cells with pale cytoplasm and dispersed chromatin


Splenic Diffuse Red Pulp Small B-cell Lymphoma
- the name kinda says it all
- a member of the splenic B-cell lymphoma/leukemia unclassifiable category (a provisional entity along with Hairy cell leukemia variant)
- involves the BM sinusoids (+/- interstitial and nodular) and PB, often with villous cytology (low level lymphocytosis, not marked like hairy-cell leuekmia variant)
- another indolent but incurable ML with responses following splenectomy
Dx'd in up to 10% of B-cell lymphomas in splenectomy specimens (spleen is usually huge)
- can see villous lymphs (like those in SMZL) in PB
- must see intrasinusoidal infiltration on BM
- spleen diffusely involved, but no follicular replacement, biphasic cytology or marginal zone infiltration
- lymphocytosis present but generally low, splenomegaly is fequent and may be massive
(+) CD20, DBA.44 IgG
(-) IgD, ANXA1, CD5/10/11c/23/25/103/123
Have low amt of IGHV gene somatic hypermutation
- lack del 7q and t(11:14), but TP53 frequently mutated
- indolent but incurable dz, splenectomy usually helps
Splenic diffuse red pulp small B-cell lymphoma

Splenic diffuse red pulp lymphoma [5] - high power shows angulated cells

Diffuse Large B-cell Lymphoma (DLBCL) Arising in the Spleen
•Variable incidence (11-34% of spleen lymphomas).
•Most cases represent secondary dissemination by systemic DLBCL.
•Primary lymphoma uncommon.
•Pts frequently present with acute fever, abdominal pain, B symptoms.
Patterns
Pattern A – Macronodular
•20/33 (61%) cases
•BCL6 positive
•Favorable outcome
Pattern B – Micronodular
•9/33 (27%) cases
•Resemble micronodular variant of T-cell histiocyte-rich large B-cell lymphoma
Pattern C – Diffuse
•4/33 (12%) cases
•Poor outcome
Splenic LBCL - Macronodular pattern




Myeloid Neoplasms
Ph* negative myeloproliferative neoplasms
•Polycythemia vera (PV)
•Essential thrombocythemia (ET)
•Primary myelofibrosis (PMF)
Chronic myeloid leukemia, BCR-ABL1 positive
Acute Myeloid Leukemia
Systemic Mastocytosis
Ph(-) Myeloproliferative Neoplasm
Prefibrotic phase
•Mild to moderate splenomegaly
•Congestion of cords and sinuses
•Platelets sequestered in spleen -> splenectomy usually contraindicated (protective effect towards thrombocytosis).
Ph(-) Myeloproliferative Neoplasm Fibrotic Phase
•Marked splenomegaly.
•Myelofibrosis; leukoerythroblastosis.
•Extramedullary hematopoiesis (EMH): expansion of RP by erythroid, granulocytic, and megakaryocytic elements.
•Splenectomy performed for cytopenias.
•Post- PV MF, post-ET MF, PMF morphologically indistinguishable
PMF – diffuse red pulp involvement

PMF

References
1. Lecture from LASOP, Jan 18, 2020.
2. Medeiros. Lymph Nodes and Extranodal Lymphomas. 2018.
3. Sohani, Aliya. Reactive Lymphoid Hyperplasia: Diagnostic Pitfalls and how to avoid them. Society for hematopathology. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=421986562
4. Elaine Jaffe. Immunohistochemical Aids for the Diagnosis and Classification of Lymphoproliferative Disorders. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=420007711
5. Gratzinger D. Spleen: Red, White, and Hungry for Blood. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=422673258
6. Cook J. Introduction to Histiocytic Disorders. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=414271529
7. YouTube video. Approach to the lymph node - Dr. Crane. https://www.youtube.com/watch?v=EbZGWXY3m-s
8. Natkunam Y. Immunodeficiency Associated Lymphoproliferative Disorders: Time for Reappraisal? Soc for Hemepath. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=412839740
9. Ponnatt T et al. Hemophagocytic Lymphohistiocytosis. Arch Pathol Lab Med. Vol 146, 4/2022.
10.