
Skeletal Muscle and Peripheral Nerve
Intro to Nerves
Acute inflammatory demyelinating polyradiculopathy (AIDP; Guillain-Barre syndrome)
Amyotrophic Lateral Sclerosis (ALS)
Spinal Muscular Atrophy (Werdnig-Hofmann disease)
Denervation Atrophy
Myasthenia Gravis
Lambert-Eaton syndrome
Duchenne Muscular Dystrophy
Becker Muscular Dystrophy
Limb-girdle muscular dystrophy
Fascioscapulohumoral muscular dystrophy
Myotonic Muscular Dystrophy (MMD)
GSD type II - Pompe's disease
GSD type III - debranching enzyme def.
GSD type V - McArdle's disease
Malignant hyperthermia (MH)
Mitochondrial Myopathies
Central core disease
Nemaline Myopathy
Centronuclear myopathy
Dermatomyositis
Polymyositis
Inclusion Body Myositis
Diabetic neuropathy
Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)
Skeletal Muscle and Peripheral Nerve
Intro to Nerves
Main components of peripheral nerves are axons and myelin sheaths (made by Schwann cells)
Somatic motor function - uses the motor unit (lower motor neuron [ant horn spinal cord]; axon to muscle; NMJ; and muscle fibers)
Somatic sensory function = distal nerve endings (can have special structures [pacinian corpuscle, etc); axon to the dorsal root ganglia; proximal nerves that connect to spinal cord or brainstem
Autonomic nerve fibers more plentiful than somatic fibers in PNS, but generally do not cause sx
Types of neuropathies:
1) Mononeuropathy - a local process involving one nerve; e.g. trauma, entrapment.
2) Mononeuropathy multiplex - a generalized but characteristically
asymmetrical process involving two or more nerves; e.g. vasculitis.
3) Polyneuropathy - generalized, multiple nerve involvement - usually distal greater than proximal and more or less symmetrical; e.g. toxic neuropathy.
4) Radiculopathy - nerve root involvement giving rise to a segmental pattern of deficit, i.e. corresponding to the innervation pattern of nerve roots (dermatomal/myotomal).
Nerve Degeneration
- regressive or destructive processes involving one or more nerve elements.
- Wallerian degeneration: axonal degeneration distal to the site of injury. Sequence of changes:
(1) early axon and myelin fragmentation;
(2) myelin phagocytosis by macrophages;
(3) Schwann cell proliferation, endoneurial fibrosis, and axonal regeneration (sprouting). Examples include trauma, ischemia (e.g. arteriosclerosis, vasculitis).
- Axonopathy: axonal degeneration occurring in a distal-to-proximal
(retrograde, or "dying back") direction. Seen in diabetes, amyloid, toxins.
- Neuronopathy: Axonal degeneration resulting from process that attacks neuron (cell body). May occur in ALS and paraneoplastic syndromes.
- Axonal degeneration is usually accompanied by myelin loss.
Onion bulb formation
- occurs in certain chronic demyelinating and/or hereditary neuropathies. It consists of a characteristic proliferation of Schwann
cells and associated collagen, in concentric layers around a central axon (in cross section resembling an "onion bulb"). This process usually occurs with repeated cycles of demyelination and remyelination and results in hypertrophy of the nerve as a whole (hypertrophic neuropathy).
Refsum disease - AR, neurologic dz c malformation of myelin sheaths around nerve cells
- has characteristic shortening of the 4th toe
- has phytanic acid storage, mostly caused by mutations in gene coding for phyanoyl-CoA hydroxylase (aka PAHX or PHYH)
- repeated episodes of myelination and demyelination result in formation of onion bulbs in Refsum dz, as well as HMSN types IB and III (Dejerine-Sottas), and CIDP
Intro to Skeletal Muscle
Muscle fibers usually distributed in a checkerboard pattern
- this patter is altered and causes muscle fiber grouping after nerve/muscle damage (denervation followed by re-innervation) after a period of months (signifying some kind of chronic condition)
- Z-bands separate the repeating units of the sarcomere; immunolabeling c B-actinin is concentrated at the Z-bands bc is made of a-actin (?)
- actin tropomyosin, and troponin found in thin filaments
- myosin and paramyosin found at thick filaments
Type I muscle: slow twitch; red fibers c lots of mitochondria and myoglobin which allows for inc oxidative phosphorylation and thus a sustained contraction (do not fatigue easily; think long-distance runners and pelagic tuna);
*** one slow red ox***
IHC: appear lighter on ATPase stain at pH 9.4; look dark blue on NADH stain
Type II muscle: fast twitch white fibers c not that much mitochondria or myoglobin (inc anaerobic glycolysis)
- will be hypertrophied c weight-training
- atrophy occurs in steroid myopathy, Cushing syndrome, collagen vascular dz, starvation, cancer, endocrine disorders
IHC: appear darker on ATPase stain at pH 9.4
Neurogenic Changes in Skeletal Muscle
a. Recent denervation - small, angulated of both fiber types. When
completely atrophied, appear as pyknotic nuclear clumps.
b. Chronic denervation (with reinnervation):
- Myofiber type grouping - random fiber type distribution is replaced by clustering of the same type, as a result of local reinnervation of
denervated muscle fibers by adjacent intact nerve twigs.
- Grouped myofiber atrophy - large groups of atrophic fibers as a result of increasing loss of motor units.
c. Seen in:
- Motor neuron diseases: ALS
- Spinal muscular atrophies: Primarily disorders of infants and children; tend to be autosomal recessive (defect in telomeric copy of SMN1 on chromosome 5q). The infantile form (Werdnig-Hoffman disease) is rapidly progressive, with death occurring by the age of 2-3 years from respiratory failure. The intermediate and juvenile forms (Kugelberg- Welander disease) progress more slowly (SMN2).
Myopathies (Primary skeletal muscle disorders)
Micro: rounding of muscle fibers on cross section, hypertrophy of fibers, increased internal nuclei, increased endomysial connective tissue, fiber splitting and ring fibers, necrosis and phagocytosis,
basophilic (regenerating) fibers, inflammatory cell infiltrates, Type 1 fiber predominance, moth-eaten and whorled fibers
- Four basic types of myopathies: Congenital, Dystrophic, Metabolic and Inflammatory.
Congenital Myopathies
- group of heriditary muscle disorders
(most autosomal dominant); usually symptomatic at birth but only slowly progressive. Characteristic morphological changes:
- "Central core" disease: Nonprogressive; infantile hypotonia; linked
to 19q13.1; assoc c malignant hyperthermia; Central light areas (cores) on trichrome (disorganized sarcomeres by EM)
"Nemaline rod" myopathy: Rod or thread-like inclusions on
trichrome; Ultrastructure: subsarcolemmal "nemaline rods" (Z-band
material)
- Centronuclear (myotubular) myopathy: Severe X-linked neonatal form; mild adult form Internal nuclei greatly increased.
Dystrophies
a number of hereditary forms of progressive and destructive myopathy chasracterized by specific gene defects; two important examples are:
- Duchenne Muscular Dystrophy (DMD): X-linked recessive disorder; onset before age 5; progressive proximal weakness,
pseudohypertrophy of calves, high skeletal muscle enzymes; death in
the 20's.
Pathogenesis: Mutations at the Dystrophin gene ("dystrophinopathy") - absence of the gene product – dystrophin.
- Becker muscular dystrophy is also a "dystrophinopathy"; later
onset around age 10-15, slower progression, less severe than
Duchenne, less often fatal.
Metabolic Myopathies
- Examples:
- Pompe's disease: Acid maltase deficiency (Type 2 Glycogen Storage Disease/ – autosomal recessive)
- Mitochondrial Myopathies with abnormal mitochondria ("ragged red fibers"); many with a maternal inheritance pattern (mitochondrial DNA) - may have cardiac and central nervous system changes as well
Inflammatory Myopathies
Lymphocytic infiltrates, fiber necrosis, regeneration
- Dermatomyositis: perifascicular fiber atrophy due to small
vessel vasculitis due to CD4-reactive T-cells. Best response to
steroids.
- Polymyositis: no perifascicular atrophy; Direct attack on muscle by CD8-reactive cytotoxic T-cells. Intermediate response to steroids.
- Inclusion body myositis
Males in 6th and 7th decade, Indolent but slowly progressive
No response to corticosteroid therapy Lymphocytic infiltration, fiber necrosis, regeneration Characteristic "rimmed vacuoles"
Granules composed of filamentous whorls by EM
? prion disorder ? (only speculation at present)
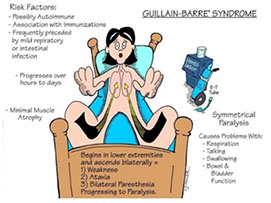
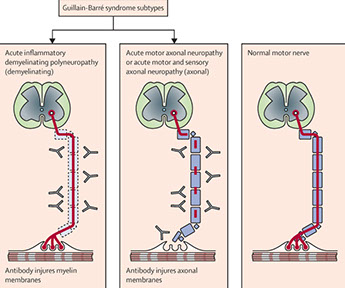
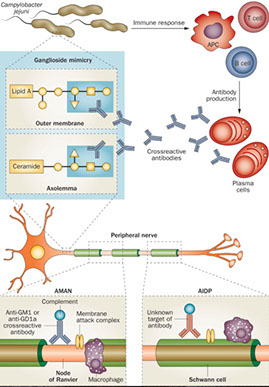
Acute inflammatory demyelinating polyradiculopathy (AIDP; Guillain-Barre syndrome)
Assoc c autoimmune attack of peripheral myelin 2/2 molecular mimicry seen c Campylobacter jejuni or herpes infx
Sx: symmetric ascending motor muscle weakness/paralysis starting in distal extremities
- 1/2 get facial paralysis and autonomic system can be affected (heart rate, BP)
Lab: albumino-cytologic dissociation in CSF (inc CSF protein c normal cell counts)
- best test is EMG / nerve conduction studies that show dec propagation of electrical impulses
Micro: endoneural inflam infiltration and segmental demyelination of peripheral nerves and motor fibers of ventral roots (sensory probs worse than motor)
Tx: must monitor pts vital capacity (for respiratory failure), may also give IVIG and do plasmapheresis
Px: nearly all pts recover in a couple of months
Chronic inflammatory demyelinative polyradiculoneuropathy (CIDP)
Segmental demyelination c relapsing-remitting polyneuropathy
Onion bulb (hypertrophic) neuropathy
Segmental demyelination
- Hereditary motor and sensory neuropathies (HMSNs)
- eg. Charcot-Marie-Tooth disease (HMSN 1) - duplication of a gene
encoding peripheral myelin protein 22 (PMP22)
- see Nervous System Non-Tumor
Amyotrophic Lateral Sclerosis (ALS)
- aka Lou Gehrig's dz
- see Nervous System Non-Tumor
Dysfunction of both UMN and LMNs with no cognitive or sensory defections that presents as weakness of unknown origin in the 20s-30s
- sporadic form is MC (familial form in 5-10%)
Sx: dec gag reflex, cough is weak, difficulty swallowing and chewing (all signs of a poor px)
- marked dec in muscle mass in prox and distal limb muscles, tongue and diaphragm
Genes: may be assoc c SuperOxide Dysmutase 1 (SOD1); or betel nut ingestion
Lab: inc CPK
Gross: atrophy of lateral corticospinal tracts and anterior motor neurons of spinal cord
Micro: Lewy like and Bunina body inclusions
- astrogliosis (sclerosis)
Tx: Riluzole (blocks glutamatergic neurotransmission by inhibiting glutamine release and inactivating voltage-gated Na+ channels)
- baclofen can treat spasticity
Px: rapid course that end in resp failure
- fronto-temporal dementia in up to 1/2 of pts
Spinal Muscular Atrophy (SMA)
AR, mutations in survival motor neutron 1 (SMN1) gene responsible for all forms
- lower motor neuron defects
- SMA1 (Werdnig-Hoffman dz) is the most severe
SMA Type I: Werdnig-Hoffman disease
MCC of SMA, presents as a floppy baby around birth (0-6 mo) and is lethal in the first 3 years, 2/2 atrophy of spinal anterior horn cells
- marked hypotonia, sluggish mvmt and fasciculation of tongue (Floppy NeoNate)
SMA II and III (Kugelberg-Welander syndrome)present later in life and have longer lifespans
Genes: Survival Motor Neuron (SMN) gene 5q11-q13
Micro: Round atrophic fibers of entire fascicles (panfascicular) w/o reinnervation
- atrophic groups of type I and type II fibers and occasionally clusters of large type I fibers
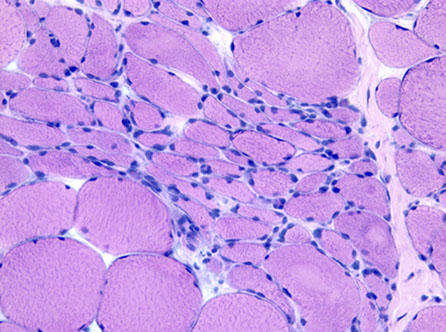
Denervation atrophy
Atrophy of sk muscle 2/2 loss of trophic innervation
- can be 2/2 loss of any part of the Lower Motor Neuron
Angular atrophic myofibers, Fiber-type grouping can be signs of re-innervation
- target fibers (nonspecific) may be seen
Myasthenia Gravis
Disorder of neuromuscular junction 2/2 ab's directed against post-synaptic AChR
- extraocular and bulbar weakness that gets worse with repetitive contractions; diplopia and ptosis
- assoc c thymic hyperplasia and thymoma
Micro: muscle bx is not informative, but can see nonspecific interstitial lymphocytes
Tx: sx can be reversed c AChE inhibitors such as edrophonium (tensilon; short-acting) and pyridostigmine (longer acting)
Lambert-Eaton Syndrome
Paraneoplastic disorder of NMJ 2/2 abs against pre-synaptic voltage-gated Ca2+ channels (resulting in dec ACh release)
- is a type II HS rxn
- improves c repetitive muscular contraction
- assoc c small cell ca of the lung
Micro: no specific pathology
Tx: no reversal of sx c AChE inhibitors alone
Duchenne Muscular Dystrophy
X-linked recessive; MC form of muscular dystrophy, onset before 5 yo
2/2 complete lack of dystrophin, which links actin to the Extracellular Matrix and can be tested (on Xp21) c IHC or Western blot
- dystrophin gene product normally functions to anchor the cytoplasm of the muscle fiber to the extracellular matrix in the dystrophin assoc protein complex
Sx: mostly males affected; normal at birth, but delayed ambulation and weakness in prox pelvic muscles; Gower's sign when getting off ground; eventually shoulders weaken and calves get pseudohypertrophied 2/2 fat infiltration
- wheelchair bound in teens and death ~20 yo, possibly from heart failure
- mild cognitive impairments
Labs: inc CK (>10k)
Micro: myopathic changes c variable fiber size, split fibers and interstitial fibrosis
- Myopathic features, including: increased variation of fiber size, increased numbers of rounded fibers and fibers with internal nuclei, muscle fiber necrosis and phagocytosis, muscle fiber regeneration (basophilic fibers), increased fibrous tissue (fibrosis) and fat (at end-stage there is little remaining muscle - replacement by fat and connective tissue).
IHC: complete lack of membranous staining c dystrophin (hence "dystrophinopathy")
Dx: dystrophin deficiency in muscle bx
Genes: XR, variety of mutations / deletions on DMD gene on Xp21.2
- can be dx's by Western blot
Tx: Roids slow the progression of DMD
Becker Muscular Dystrophy
2/2 functional mutations or partial loss of dystrophin protein
- X-recessive, less severe with later onset and normal life-span
Calf hypertrophy is an early finding
- able to walk beyond 15 yo
- MR is not as common as in DMD
Labs: CK levels and muscle bx is similar to DMD
- Western blot shows low dystrophin
- genetic testing is positive for mutation in 65% (same as in DMD)
IHC: weak and patchy dystrophin staining
Limb-girdle Muscular Dystrophy
AR, M=F, group of dzs 2/2 abnormalities of components of the dystrophin-associated protein complex other than dystrophin
- usually affects the shoulders and pelvis
Labs: CK elevated 10x ULN and remains elevated
Fascioscapulohumeral Dystrophy
AD, seen in early teens c asymmetrical muscle wasting on back, shoulders and upper arms
Labs: CK is 2x ULN and remains elevated
Myotonic Muscular Dystrophy (MMD)
MCC muscular dystrophy in adults, AD, 2/2 CTG trinucleotide expansion in Dystrophia Myotonia Protein Kinase (DMPK) gene on 19q in type 1 and cr 3q in type II and shows anticipation genetically
Sx: smooth muscle effects such as bowel dysmotility, myotonia (prolonged contractions), cardiomyopathy, and nonmuscular features like cataracts, "hatchet facies," (facial atrophy and temporal balding), DM
Micro: markedly inc # of internalized (centrally placed) nuclei, sarcoplasmic masses, and ring fibers are characteristic
- moth-eaten and whorled fibers can be seen
Pompe disease
- aka Glycogen storage disease (GSD) type II
Mutation in gene encoding a1,4-glucosidase (acid maltase) causes lysosomal accumulation of glycogen that affects the heart
Forbes / Cori dz (Debranching enzyme deficiency)
- aka GSD type III
accumulation of unbound glycogen, esp in liver
McArdle's disease
- aka GSD type V
2/2 absence of myophosphorylase
Sx: exercise-induced cramps and rhabdomyolysis c unbound glycogen accumulations
Malignant Hyperthermia (MH)
syndrome of malignant hyperthermia when exposed to certain anesthetics
- hypercarbia while under anesthesia is often the 1st sign
- caused by defective RYR1 calcium channels due to RYR1 gene mutation
Mitochondrial myopathies
Maternal inheritance; commonly see lactic acidosis, several subtypes
- Mitochondrial encephalopathy, lactic acidosis, and stroke-like symptoms (MELAS)
- Myoclonic epilepsy and ragged-red fibers (MERRF)
- Kearns-Sayre syndrome (KSS): opthalmoplegia, cardiac conduction defects, sensorineural hearing loss
- Chronic progressive external opthalmoplegia (CPEO)
Micro: "ragged red fibers" on trichrome, which have accumulations of glycogen and neutral lipids and look red c red dotted sarcoplasm 2/2 mitochondrial prolif
IHC: inc reactivity succinate dehydrogenase (SDH)
- dec reactivity for cytochrome c oxidase
EM: paracrystalline inclusions
Central core disease
Nonprogressive; infantile hypotonia; linked to 19q13.1; assoc c malignant hyperthermia when given inhaled anesthetics
Ryanodine receptor gene (RYR1) mutations --> inc risk malignant hyperthermia
Histo: centrally disorganized myofibrils
Central light areas (cores) on trichrome (disorganized sarcomeres by EM)
Nemaline myopathy
Nemaline rods (derived from Z-band) accumulate
Rod or thread-like inclusions on trichrome;
- affects sk muscle of face, neck, and limbs, causing difficulty swallowing, foot deformities, scoliosis, and joint contractures
Genes: NEB gene mutations (~1/2), ACTA1 mutations (up to 1/4)
Micro: nemaline rods best seen as dark blue structures c Gomori trichrome stain
EM: subsarcolemmal "nemaline rods" (Z-band material), a dense linear structure that appears to be derived from Z-lines
Centronuclear (myotubular) myopathy
Severe X-linked neonatal form; mild adult form Internal nuclei greatly increased.
Micro: Accum of fibers c chains of central nuclei that look like the myotubular stage of muscle development
Dermatomyositis (DM)
Inflammatory myopathy c generalized malaise and prox weakness
- Gottron's papules and heliotrope rash characteristic
- adults have inc risk of visceral malignancy (inc 40%)
- kids get calcinosis and myocarditis and visceral ischemia (juvenile dermatomyositis)
Labs: inc CK, aldolase, (+) ANA, anti-Jo-1
Micro: ab mediated damage to BV's results in perifascicular inflam/atrophy; may also see lymphocytic vascullitis
*** the perifascicular inflam is closer to the skin, which gives it the skin sx (not seen in PM, which is endomysial)***
EM: tubuloreticular inclusions (means interferon is inc)
Tx: roids
Polymyositis (PM)
Autoimmune dz, mostly adults, F>M; presents c inc CK and aldolase (in 2/3), myalgia, symmetric proximal weakness
- can distinguish from dermatomyositis bc does not have rash, or papulles
- not assoc c inc risk of malig or calcinosis
Micro: cell-mediated invasion of viable myofibers by CD8>4 lymphs --> endomysial inflam
- no vasculitis and no perifascicular atrophy
Tx: roids, interferon
Mixed Connective Tissue Disease
Autoimmune disorder that has features of three other connective tissue diseases:
-SLE
-Scleroderma
-Polymyositis
Sx: Raynaud phenomenon, sausage-like fingers, inflammed joints + muscles, pulmonary hypertension
Tx:Corticosteroids, Hypertension drugs
Labs: CK and Aldolase increased
Inclusion Body Myositis (IBM)
Similar sx as polymyositis; but MC in males >50 yo, prominent involvement in finger/wrist flexors, no response to roids
- etiology generally unknown, but some cases can be attributed to Valosine Containing Protein (VCP) gene mutations
- rimmed vacuoles not seen on paraffin embedded sections of skeletal muscle and need frozen and/or glutaraldehyde fixed material for plastic embedding and possibly EM
- frozen material larger than resin-embedded and less apt to be negative due to sampling
Micro: tubulofilamentous inclusions, inclammation, rimmed vacuoles and fibers c mitochondrial abnormalities
Tx: resistant to roids and immunosuppression; usually go straight to methotrexate
Px: relentlessly progressive, fatal disease
Diabetic Neuropathy
Seen in about 1/2 of diabetics, causing distal sensorimotor disturbances ("stocking glove distribution" of numbness)
Micro: loss of axons, thickening of small BVs and perineurium
Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)
Prolonged clinical course; sensory and/or motor neurons can be involved and it is usually symmetric
Micro: concentric Schwann cell lamellae ("onion bulbs")
Tx: roids may be beneficial



Refsum dz, shortening of the 4th toe




Denervation atrophy - affects both muscle types


Here, GBS is fairly acute, and nerve contains inc inflam. Majority of small round nuclei are lymphs infiltrating nerve; residual myelinated axons can be seen. Denser pink lines (black arrow) are axons and bubbly-appearing pink areas surrounding them are myelin sheaths.
Small onion bulbs

SMA type 1: Werdnig Hoffman dz


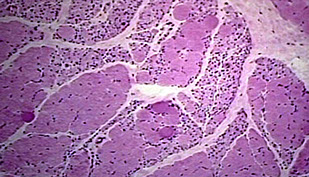


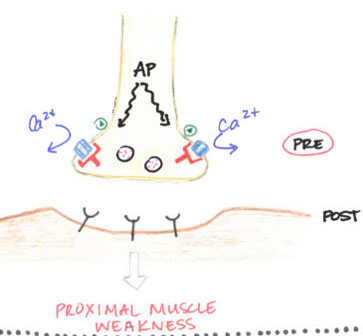
Lambert-Eaton syndrome: Abs against presynaptic Ca2+ channels; weakness improves c muscle contraction



Duchenne muscular dystrophy




Mitochondrial myopathies
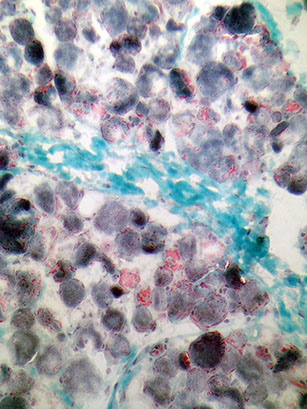
Central core disease


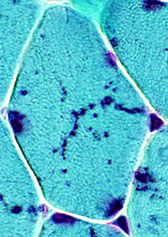
Nemaline myopathy
Gottron's papules
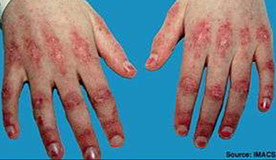

DM

Polymyositis (PM)

IBM

