Pharmacology
Pharmacokinetics
- CLADME process
Volume of Distribution
Half-life
Steady state
Therapeutic Drug Monitoring (TDM)
Methods for TDM
- lidocaine
- methotrexate
Pharmacokinetics
Mathematically describes the fate of the drug after administration in a given dosage form and a given route of administration.
Compares a drug with other drugs or compares a drug dosage form with other dosage forms
Predicts blood levels of a drug with different dosage regimens or predicts blood levels of a drug with different disease states
Factors affecting pharmacokinetics
Age
Gender
Genetics
Diet
Health habits
Pathology
CLADME Process
Compliance
Take the drug appropriately
Responsibility lies with:
Physician, Pharmacist, Patient
Non-compliance includes:
Taking drug more or less prescribed
Skipping doses
Taking for shorter lengths of time
Steady state must be achieved and maintained
Adolescence, genetics, complications from other drugs, metabolism
Administration
Enteral
Oral – most common route of administration
Sublingual – Placement under the tongue
Rectal – 50% of the drainage in the rectal region bypasses the liver which will prevent the destruction of the drug by intestinal enzymes.
Perenteral - used for drugs that are poorly absorbed from the GI tract
IV – most common parenteral route
IM – used for drugs that dissolve slowly
SC – absorption is slower than the IV route
Liberation
Oral doses must be released from the dosage form and made soluble
Rate of release depends on:
Drug formulation
Dosage
Ionization state
Un-ionized form readily pass membranes
pH of the environment
Protein binding
Proteins have the ability to bind drugs
Non-protein bound drugs
The degree of protein binding varies from drug to drug
It is consistent between individuals for a given drug
Varies with age
Drugs will exist in blood in equilibrium between the bound and the free form
Free form drugs are the active form
Conditions altering the binding protein concentrations, such as albumin, greatly affects the amount of free drug
Endogenous substances may also compete with binding sites on proteins and displace a given drug
Absorption
Describes the rate and degree to which a drug leaves the site of administration and enters the circulation
EXCEPTION: IV administration of drugs
Here the rate depends on
Physical & chemical characteristics of the drug (solubility & pKa)
Local physiology (pH, perfusion rate, GI motility)
Polarity
Oral administration
Mouth – stomach – intestinal lumen – portal circulation for “first pass metabolism”
Distribution
Delivery of drug via circulation
Depends on:
Binding of protein
Transportation
Ability to cross cellular membranes
Rate of tissue perfusion
Lung
Skeletal muscle
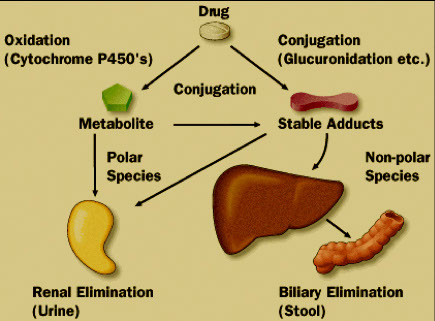
Metabolized
Most takes place in the liver
Uses Cytochrome P450
Primary purpose is to transform drug into its polar or water soluble form
Metabolites are formed
Occurs in phases
Phase 1
Phase 2
First-order (linear)
The amount of metabolite produced steadily increases in proportion to an increasing amount of parent drug in the system
The rate of metabolite is a constant percentage of parent drug eliminated per unit of time
Examples: antibiotics and most other drugs
Zero-order (non-linear)
The rate of metabolism approaches Vmax
The rate of metabolite formed is constant and independent of the amount of parent drug eliminated per unit of time
Example: alcohol
Excreted
Elimination occurs in a variety of ways
- Filtration
- Secretion
- Reabsorption
How a drug is eliminated dictates:
Monitoring drug
Monitoring metabolite
Volume of Distribution
A hypothetical volume of fluid into which the drug is disseminated
Relates the volume of drug in the body to the concentration of drug in the blood
Process takes between 30 minutes and 2 hours
C = D/Vd or Vd = D/C, where
- C = plasma concentration of drug
- D = Total amount of drug in the body
Example:
If 25 mg of a drug (D=25mg) is administered and the plasma concentration is 1.0 mg/L, then Vd = 25 mg/1.0 mg/L = 25 L
Half-life
Definition:
time needed for the serum concentration to decrease by one-half
Formula for calculating:
t½ = 0.0.693 Vd/total body clearance (TBC)
TBC = clearance rate /mass
Example:
What is the half-life of a drug, which had a Vd of 1.1 L/kg and a clearance of 15 ml/min in a patient weighing 34 kg?
t½ = 0.0.693 Vd x 1/TBC
t½ = 0.693 (1.1 L/kg x 1min/15mL x 1000mL/L x 34 kg)
t½ = 1728 min or 29 hrs
Steady state
Occurs when ?:
the rate of drug elimination is equal to drug administration
Occurs how ?:
multiple doses are given to an individual
Drug accumulates in the body until
drug elimination = drug administered
Multiple doses given at the intervals equal to the drug’s half-life will result in Steady state
This is usually achieved after 6 or 7 half-lives after initial dosing
Therapeutic Drug Monitoring
Serial, routine testing for dose optimization
Blood (plasma, serum, whole blood)
The drug(s) to test for is known
Related and often predictable
Blood concentrations
Pharmacokinetics: What the body does to the drug
Pharmacodynamics: What the drug does to the body
TDM Medications: meet 1+ of these criteria
Narrow therapeutic index
Used for long-term therapy
Correlation between serum concentration and clinical response
Wide variability in individual patient PK
Absence of a biomarker associated with a therapeutic outcome
Administered with other, potentially interacting compounds
When is TDM needed??
Suspected drug overdose
Unknown medication in comatose patient
Dose optimization in critically ill patient
Toxic effects look like disease symptoms
Disease state alters pharmacokinetics
Lack of therapeutic effect
Compliance issues
Prophylactic drug
Drug interaction/multidrug therapy
High does Methotrexate (Leucovorin rescue)
When to measure free drug concentration??
Highly protein bound drugs (>90% bound)
Recognize what has been dosed is far greater then what is immediately available
Protein concentration is very IMPORTANT
CAUTION:
Albumin < 2.5 g/dL
Renal Failure
Pregnancy
Age Extremes
ANY TIME PROTEIN STATUS IS ABNORMAL BE CAUTIOUS
When to measure metabolites???
Metabolically active metabolites
Primidone
Phenobarbital
Carbamazepine
Procainamide
Total concentration of drug and active metabolites important
TCA’s (Amitriptyline and Nortriptyline)
CAUTION: Immunoassays may cross react with inactive metabolites
Avoid gel separator tubes for collection of specimens intended for drug analysis
Why? - because the drug may diffuse from the serum into the gel, causing a reduction in measured drug level.
Evaluate other conditions of specimen collection and handling
Preservatives
Heat
Light
Freeze/thaw
Establish critical values
Report results with therapeutic ranges
Report toxic thresholds
Methods for TDM
Automated Assays
Primarily immunoassays
Limited to common drugs
LC-MS/MS
Reference Labs
Assays and TAT vary widely
Troubleshooting - what can go wrong
Is the value accurate
Test properly validated
QC appropriate
Does the value make sense
Is it compatible with life?
Is it unexpected?
Reasons for spuriously high or low values
Phlebotomy error
Specimen mix up
Change in patient status
Drug history
Change in formulation or manufacturer
TDM in organ evaluation
Drug is metabolized by a particular organ
Requires specific metabolism
Decreased production of metabolite:
Organ not functioning
OR
Blood flow to organ is interrupted
ex:
Lidocaine to measure liver function
Method used after transplant or in critically ill patient to check liver function
- P450 in liver turns lidocaine into Monoethylglycinexylidide (MEGX)
Method:
1. Measure MEGX level
2. Bolus lidocaine
3. Measure MEGX level
Low MEGX production suggests:
Diminished blood flow
Metabolism decreased
Liver failure
P-450 system not working
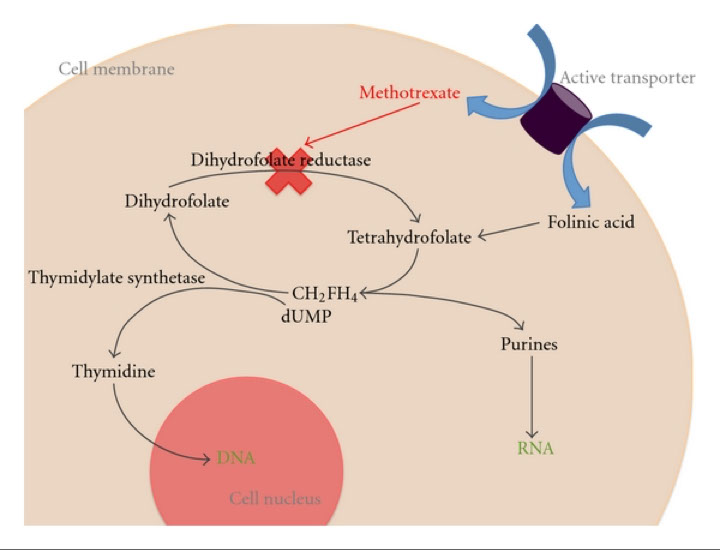
Methotrexate: Dose detemines function
Immunosuppression <10nM
Psoriasis
Refractory Rheumatoid arthritis
Cancer Chemotherapy 50nM
Ordinary dosing
Very High dosing
500 times ordinary dosing
Inhibits dihydrofolate reductase
Shuts down DNA synthesis
Leukovorin rescue
In cases of very high methotrexate dosing
Leukovorin
N-5-formyltetrahydrofolate
Product of dihydrofolate reductase
Leukovorin infused 18-36 hours after methotrexate
Methotrexate levels must be monitored