
Nervous System Tumors
Introduction
Pineal Region Tumors - Perinaud's Syndrome
Histology
-
the Cells
-
Neurons
- Pyramidal neurons, cortical granular (stellate) neurons, Betz cells, Purkinje cells, anterior horn cells
-
Glia
- Astrocytes, oligodendrocytes, ependyma, choroid plexus, microglia
-
Neurons
- Cerebrum, Hippocampus, Thalamus, Basal ganglia, Amygdala, Cingulate gyrus, Midbrain, Pons, Medulla, Cerebellum, Pineal gland, Pituitary gland
Pineocytoma
Pineal Parenchymal Neoplasm of Intermediate Differentiation
Pineoblastoma
Sellar Region Tumors
Pituitary Adenoma
Craniopharyngioma
Other causes of Sellar Region Masses
Primitive Tumors: Small Round Blue Cells
Meduloblastoma/Primitive Neuroctodermal Tumor (PNET)
Atypical Teratoid Rhabdoid Tumor (AT/RT)
Astrocytic Glial Tumors
Clival Tumors
Pilocytic astrocytoma
Subependymal Giant Cell Astrocytoma (SEGA)
Angiocentric Glioma
Infiltrating Astrocytoma
Pleomorphic xanthoastrocytoma (PXA)
Anaplastic astrocytoma
Glioblastoma
Oligodendrocytic Glial Tumors
Oligodendroglioma
Anaplastic oligodendroglioma
Mixed gliomas
Chordoma
Chondrosarcoma
Giant Notochordal Hamartoma
Ecchordosis Physaliphora
Dural-Based Tumors
Meningioma
- Menngothelial, Fibrous, Psammomatous, Secretory, Atypical, Clear cell, Choroid, Papillary, Rhabdoid, Anaplastic
Hemangiopericytoma
Meningioangiomatosis
Ependymal Glial Tumors
Subependymoma
Myxopapillary Ependymoma
Ependymoma
Clear cell ependymoma
Anaplastic ependymoma
Brain Cysts
Rathke's Cleft Cyst
Epidermoid cyst
Colloid cyst
Enterogenous cyst
Cysticercosis
Choroid Plexus Tumors
Strange Tumors of the CNS
Choroid plexus papilloma
Atypical Choroid Plexus Papilloma
Choroid plexus Carcinoma
Ganglioglioma
Desmoplastic infantile ganglioglioma/astrocytoma
Dysembryoplastic Neuroepithelial Tumor (DNET)
Hemangioblastoma
Choroid glioma of the 3rd ventricle
Paraganglioma
SpinoCerebellar Ataxia type III (SCA3) / Machado-Joseph disease
Calcifying Pseudoneoplasm of Neuroaxis (CAPNON)
Rare Neurocytic Tumors
Central Neurocytoma
Gangliocytoma
Dysplastic Gangliocytoma of the cerebellum (Lhermitte disease)
Peripheral Nerve Sheath Tumors
Ganglioneuroma
Ganglioneuroblastoma
Neuroblastoma (esthesioneuroblastoma) - also see Adrenal
Neurofibroma
Malignant Peripheral Nerve Sheath Tumor
Schwannoma
Traumatic neuroma
Perineuroma
Neurilemmoma of VIII CN - see Ear
Introduction
Histology
The cells
Neurons
Pyramidal neurons - found in the cortex and parts of the hippocampus, these big, triangle-shaped cells c prominent nucleolus which have big apical dendrite that shoots towards the surface of the brain (in case you get disoriented)
Cortical granular (stellate) neurons
Granular neurons - seen in the dentate fascia of the hippocampus, as well as the cerebellum and form a "molecular layer" 2/2 their extensive dendritic processes
Betz cells
Purkinje cells
Anterior horn cells
Glial Cells
Astrocytes - star-shaped 2/2 radial processes which have GFAP; should be about 10x as numerous as neurons in an adult brain
- their foot processes help to form the blood-brain barrier, relation to Aquaporin4 (?)
- proliferate if neurons are lost ([astro-] gliosis)
-
Oligodendrocytes
Ependymal cells - Choroid plexus
Microglia
Resident tissue histiocytes of the brain.
- bc they are histiocytes, they are CD68+
Cerebrum
The outer layer is the gray matter, the infoldings are sulci; there are 6 layers of the cerebrum (1. outer plexiform (molecular) layer; 2. outer granular layer; 3. outer pyramidal layer; 4. inner granular layer; 5. ganglionic/inner pyramidal layer; 6. multiform cell layer)
- layers 3 and 5 are larger bc their axons go to other parts of the CNS, sometimes all the way to the spinal cord
- the pyramidal neurons point towards the surface
- the little space underneath the CA (looks like edema) is normal
The neonatal cerebral cortex comes from neuroblasts that spread out from the germinal matrix. The germinal matrix lasts until about termt
Hippocampus
The hippocampal formation = hippocampus, dentate gyrus, and subiculum and is assoc c entorhinal cortex (which feeds into the hippocampus)
- the "Napolean hat"-shaped Lateral Geniculate Nucleus is close by
Subiculum is at base of hippocampus and has pyramidal neurons and becomes cornu ammonis (CA)-1, then CA2, CA3 and finally wrapping around into the hilum
- the little squiggly of cells that separates the hilum and the subiculum is the dentate fascia
- CA3 has the largest pyramidal neurons, CA2 is the thinnest, CA1 will get messed up yo in hypoxia
- stratum granulosum - tightly packed smaller granular cells that make up the dentate fascia (C-shaped part of the dentate gyrus)
- the alveus, bwt CA neurons and the lateral ventricle, feeds into Papez circuit (alveus --> fimbria hippocampus --> fornix --> hypothalamus and mamillary bodies)
Amygdala - disorganized blob of cortex
Layer 2 pre-Alpha clusters - anterior portion of hippocampus; entorhhinal cortex, should find lots of plaques and tangles
Tela choroidea - 3 cell layer loose portion of highly vascular connective tissue that gives rise to the choroid plexus
Thalamus
Has multiple nuclei, coordinator of sensory information
- no big difference histologically bwt different parts, but should see larger neurons that project their axons around the brain and smaller regulatory neurons that inhibit signals c GABA
- football-shaped nucleus = subthalamus
- may also see some of the substantia nigra pars compacta there, depending on the cut
Basal Ganglia
Includes the caudate, putamen, and globus pallidus
- 19/20 of the neurons are inhibitory (GABA-inergic) that go to the globus pallidus from the caudate, putamen and nucleus accumbens
- Fahr's (extinct disease) - once that that calcifications caused dz
The pencil fibers of Wilson are white matter bundles found in the gray matter neuropil
Amygdala
Group of nuclei covered by entorhinal cortex, looks generally disorganized
- functionally related to the pyriform lobe
- has some control over the hypothalamus
- ablation of the amygdala may result in Kluver-Bucy syndrome (hypersexual, hyper-oral, visual agnosia, decreased fear)
Cingulate Gyrus
Midbrain
A good slice of midbrain will have the cerebral peduncles in the anterior portion which contain the crux cerebri
- posterior to the crux are the substantia nigra with their darkly-staining cells; the red nuclei (which help control gait and are red 2/2 iron from hemoglobin and ferritin) are posterior to the substantia nigra
- the oculomotor nerve leaves anteriorly through peduncles, but the oculomotor nuclei are more posterior, found anterior to the cerebral aqueduct
- in the most posterior part of midbrain are the superior colliculi (makes sense that they are on the same slice as the oculomotor nuclei)
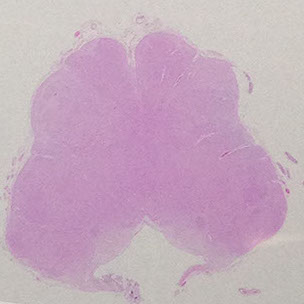
Pons
Has a posterior tegmentum assoc c the 4th ventricle and an anterior base with crossing pontocerebellar fibers (that make up the middle cerebellar peduncle)
- the pigmented locus ceruleus, which makes NE, is lateral to the 4th ventricle
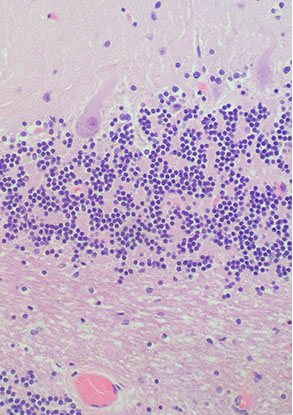
Medulla
Forms the floor of the 4th ventricle and has CNs VIII-XII
- the inf medulla has the pyramidal tract anteriorly and the inf olivary nucleus
- arcuate nucleus in the anterior portion as well
- dorsal motor nucleus of vagus one of the first places to get Lewy bodies in Lewy body dz
Cerebellum
From out to in, the cerebellum is made of the molecular layer, the Purkinje cell layer, a granular cell layer and an inner tract of white matter
- Purkinje cells are large neurons that get info from the granular layer and send it out into the
- Bergman glia also live im the Purkinje cells layer
- glomeruli are little pink structures found in the granular layer
*** Dogs Eat Great Food ***
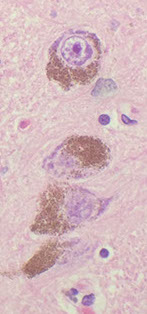
Pineal gland
Produces melatonin (inhibited by light which goes through retina to suprachiasmatic nuclei to pineal), functions in sexual maturity, circadian rhythm, antioxidant
- found in subarachnoid space below splenium of corpus callosum above superior colliculi
Has pinealocytes (make melatonin and secretes into pineal capillaries)
- may be calcified (inc c age)
- can also get cysts and tumors, which can obstruct aqueduct of Sylvius
Pituitary gland
Darker anterior lobe (adenohypophysis) derived from Rathkes pouch (oral ectoderm), and smaller, lighter posterior lobe (neurohypophysis) developed from hypothalamus infundibulum (still connects by stalk of pituitary to hypothalamus, the pathway for oxytocin and vasopressin)
- found in the sella turcica of sphenoid bone, can be larger in preg and multiparous women from hyperplasia (inc vascularity)
- irrigated by internal carotid artery by sup and inf hypophyseals
-- arterial blood goes to posterior lobe and venous blood to the anterior lobe by the portal system (anterior lobe vulnerable to hypoperfusion in case of shock
*** B-FLAT - Basophils, FSH, LH, ACTH, TSH ***
*** GPA - GH, Prolactin, Acidophils ***
- anterior lobe has chromophils (acetophils, basophils) and chromophobes (don't secrete anything)
-- acetos and basos make all the hormones (except the "trophs" [somatotrophs, lactotrophs, thyrotrophs, corticotrophs, and gonadotrophs])
- also intermediate lobe, which looks like thyroid
- posterior lobe looks like brain ... because it is brain
IHC: (+) nested reticulin positivity
Pituitary gland can be aplastic or hypoplastic (assoc c anencephaly)
- can have a pharyngeal pituitary, which is a small rest of ectopic pituitary
Pituitary apoplexy is hemorrhage and necrosis in case of pituitary macroadenoma (or other enlargement)
- Sheehan syndrome is postpartum pituitary necrosis in the anterior pituitary from hypovolemic shock after being physiologically enlarged during pregnancy (the posterior pituitary is not affected bc of the different blood supply)
- need at least 90% destruction of the anterior pituitary for hypopituitarism
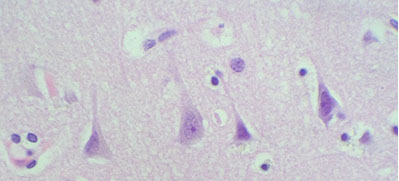
Pyramidal neurons, pointing to the surface

Hippocampus - granular neurons in dentate fascia with adjacent molecular layer
Molecular layer
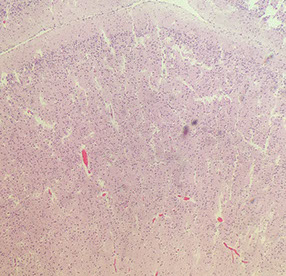
Neonatal cerebral cortex
Cerebral cortical layers
White matter
Molecular layer
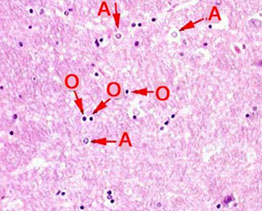
Astrocytes (A) and Oligodendrocytes (O)

Sulcus -->
Gray matter
White matter
Cerebrum

Microglia, CD68

H
CA3
CA2
CA1
Sub
DF
Hippocampus
CA = cornu ammonis; H = hilum; Sub = subiculum; DF = dentate fascia (alveus not visible, but should be somewhere around CA1)
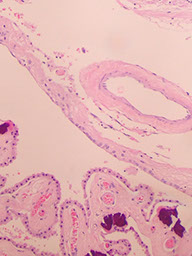
Tela --> choroidea

Thalamus
Basal ganglia - Pencil fibers of Wilson (arrow)
-->
Midbrain (R = Red nuclei; SN = Substantia nigra)
CN III
<-- cerebral aqueduct
superior --> colliculi
<-- SN
R
R -->
Peduncle
Anterior
Midbrain - Sub nigra

Pons
4th Vent
Locus ceruleus
Medulla
P
H
V
O
P = Pyramids; O = Inf olivary nuclei; H = hypoglossal nucleus; V=vestibular nuclei)

4th ventricle
crossing pontocerebellar fibers
tegmentum
|
|
|
base
|
|
|
|
|
|
|
|
|
|
|
Anterior
Pons
A
A = abducens nuc
Cerebellum - Molucular layer
Granular cell layer
<-- Purkinje cell
White matter
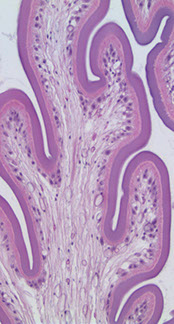
Pineal gland
Pineal gland

Cerebellum
M
P
G
W
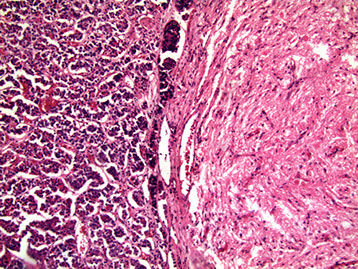
Neurohypophysis
(posterior)
Adenohypophysis
(anterior)
Astrocytic Glial Tumors
Pilocytic astrocytoma (PA)
MC pediatric brain tumor, often found as a cyst in cerebellum, thalamus, optic pathway and spinal cord; Grade I
Imaging: Cyst (pretty well-circumscribed) with enhancing mural nodule, or may be a solid enhancing mass
- first form small cysts, which then fuse to form larger cysts
- can be in optic nerve region in someone c NF1
Micro: Non-infiltrating glioma comprising a proliferation of bland, "hair-like" (mildly atypical bean-shaped) nuclei with bipolar hair-like processes
- low power shows biphasic architecture with microcystic and fascicular (compact c stretched-out cells c thick processes) areas
- Rosenthal fibers (eosinophilic elongate structures) and eosinophilic granular bodies (EGB) are frequently seen
Perivascular chronic inflam is common
Can see "degenerative" nuclear atypia; no mits
- does not infiltrate and Ki-67 is low (<2.5%)
Must distinguish from reactive gliosis surrounding a brain lesion ("piloid gliosis") which looks very similar (also with Rosenthal fibers, and more...)
Need mural nodule and not cyst wall to dx
- almost anything can cause Rosenthal fibers as a result of compression
Genetics: BRAF gene (7q34) tandem duplications cause several KIAAI549:BRAF gene fusion in >1/2 of PAs
BRAF gene (7q34) point mutation (V600E) if supratentorial (mostly b9 tumors)
– Pleomorphic xanthoastrocytomas (66%)
– Diencephalic pilocytic astrocytomas (30%)
– Gangliogliomas (20%)
DDx: hemangioblastoma and suprasellar craniopharyngioma (can have Rosenthal fibers in gliotic cyst wall)
- eosinophilic granular bodies (EGBs) can be seen in: 1) PA; 2) PXA; 3) Ganglioglioma
- Cyst with a mural nodule in the cerebellum of an adult?
= Hemangioblastoma!
Px: Rarely progress to anaplastic subtypes
- fairly b9
Subependymal Giant Cell Astrocytoma (SEGA!!!)
Assoc c tuberous sclerosis (>9/10); WHO grade I
Found in medial wall of lat ventricles, may block formen of Monroe, causing hydrocephalus
- also has strong assoc c epilepsy (remove to stop seizures)
- develop in first 2 decades of pts c tuberous sclerosis
- not just an astrocytome, also has neural and glial cells
Imaging: Variable enhancement (not ring) may show calcifications
Micro: pleomorphic prolif of bizarre "confused cells" - mixed neuronal and gangliocytic differentiation
- although the cytoplasm appears astrocytic (homogenous, eosinophilic), the nuclei often have neuronal features (granular chromatin, prominent nucleoli)
- "confused cells" can also be seen in cortical dysplasia
Although may see lots of nuclear pleomorphism, there are no mits and Ki67 is low (looks bad, but has a good prognosis)
- tumor often has dystrophic calcifications
Cave canum: this tumor is rare and may be overcalled as a glioblastoma
DDx: subependymoma, central neurocytoma, choroid plexus papilloma, pleomorphic xanthoastrocytoma
IHC: Block (+) for S-100, (+) GFAP (glial component), and NFP (in neural component)
Angiocentric glioma
Recently described (rare...)
Seen in kids and young adults; causes seizures
Localized to the cerebral hemispheres
Microscopically see low-grade infiltrating glioma with astrocytic nuclear features
- key is seeing the tumor cells wrapping around the outside of blood vessels (angiocentricity)
(+) GFAP
Shares phenotypic overlap with ependymoma (dot-like perinuclear activity)
Granular astrocytoma
Lysosome-rich cells c peripheral staining c GFAP and are pos c both CD68 and PAS can lead to confusion c histiocytes
- careful exam shows background histiocytes which are smaller than the tumor cells and are more intensely positive
- this variant is bland histologically, but has an aggressive course
Pilomyxoid astrocytoma
First separated from pilocytic astrocytoma in 1999
- present in young pts (<1 yo)
- predominance in hypothalamus / chiasm, though can be seen elsewhere
- like pilocytic astrocytoma they are well-circumscribed and enhance on scans
Micro: similar histology as pilocytic astrocytomas c myxoid background and bipolar cells
- cells radiate around vessels, do not have Rosenthal fibers and eosinophilic granular bodies
Px: are more aggressive than pilocytic astrocytomas, and considered WHO grade II
Infiltrating Astrocytoma
WHO grade 2; Infiltration of brain parenchyma; white matter derived, and expands white matter
Seen in young adults (in their 30s), cerebral hemispheres MC site
Abnormal FLAIR/T2 signal with mass effect, usually in white matter
- No contrast enhancement!
Micro with scattered (often hypocellular) infiltrating atypical astrocytic nuclei
- atypia is enlargement hyperchromasia, nuclear membrane irregularities (benign is smooth)
No mits, necrosis, or microvascular prolif
May see neoplastic cells with prominent eosinophilic nuclei (gemistocytes [stuffed cells])
- Neoplastic gemistocytes usually mean worse prognosis
Pleomorphic xanthoastrocytoma (PXA)
Superficial temporal lobe tumor assoc c seizures in kids and young adults; xantho = lipidized
On imaging is a superficial cyst with an enhancing mural nodule in the temporal lobe (supratentorial)
Micro: highly atypical appearing (yet mostly benign) tumor with atypical giant cells c astrocytic nuclear features (striking pleomorphism) that look like they have cytoplasmic lipid vacuoles (thus "xantho")
- eosinophilic granular bodies (EGBs) can be seen in: 1) PA; 2) PXA; 3) Ganglioglioma
Mits and Ki67 are low/rare despite the pleomorphism
- reticulin stain may highlight the increased pericellular reticulin deposition
IHC: (+) pericelllular reticulin positivity, GFAP
- If this kind of tumor shows lots of mits and high Ki67, it is a WHO grade III, otherwise is a WHO grade II
BRAF gene (7q34) point mutation (V600E)
– Pleomorphic xanthoastrocytomas (66%)
– Diencephalic pilocytic astrocytomas (30%)
– Gangliogliomas (20%)
Tx: excision
Anaplastic astrocytoma
Seen as an abnormal T2/Flair signal with mass effect and patchy contrast enhancement in middle-ages adults
- has same microscopic features as infiltrating astrocytoma but with increased cellularity, more atypia and more mits (which is why it is a WHO grade III)
Pediatric High Grade Glioma (HGG)
Genes:
Diffuse Intrinsic Pontine Glioma (DIPG)
Most severe pediatric solid tumor
Genes: somatic H3-K27M and / or H3K27 trimethylation
- H3K27M is a histone protein
Tx:
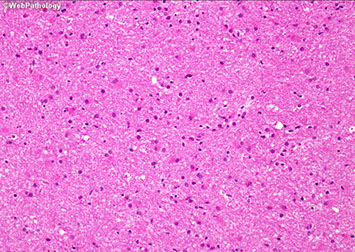
Glioblastoma multiforme (GBM)
More common in older pts (50's), involving the white matter
Imaging: heterogenous ring-enhancing mass with surrounding T2/FLAIR signal abnormality; if it crosses the midline, very likely to be a glioblastoma (butterfly lesion classic)
Micro shows an atypical infiltrative astrocytic glioma with frequent mits, pleomorphism, microvascular prolif and pseudopalisading necrosis
- either necrosis or microvasc prolif req'd for dx
- microvascular prolif is not an increase in the number of vessel spaces, but an increase in the thickness of the cellular coat of pericytes
-- thus to see microvasc prolif a smooth muscle actin (SMA) helps but a CD31 does not
Primary glioblastomas (de novo) occur in older pts, have EGFR amplification and/or PTEN mutations and have a very poor px
Secondary glioblastomas arise from a previous tumor that mutates and overexpresses p53, and has IDH1 mutations
- these have a slightly better px
- p53 usually seen in glial tumors (not as much in oligodendrogliomas) although p53 can be negative depending on the molecular pathway the tumor has taken
Genes: Isocitrate Dehydrogenase 1 mutation (IDH1): point mutation (R132H) in >1/2 of astrocytomas, oligodendrogliomas, and "mixed" gliomas (WHO Grades II-III)
- found in nearly all oligodendroglioma
- the mutation conveys a better px (wild type is worse)
-- mutation found in secondary GBM (since it is retained from its primary non-GBM tumor type) but NOT in primary GBM
- pts c hypermethylation of O(6)-methylguanine-DNA methyltransferase (MGMT) are more responsive to temozolomide and radiation tx
- higher expression of PD-L1 assoc c poor px [1]
- there are some treatments for blocking mutant IDH1 strains
Turcot syndrome
Type 1 = HPNCC (Lynch syndrome) + GBM
- mutated mismatch repair genes (MLH1, MSH2, MSH6, PMS2)
Many different subtypes (small cell, giant cell...)
- Epithelioid GBM found to have BRAF V600E mutations
For differentiating treatment effects vs a recurrent GBM, Pseudopallisading necrosis is found in an actively growing glioblastoma
- pseudopallisading necrosis is not a result of treatment
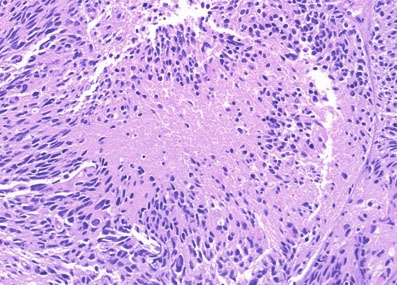
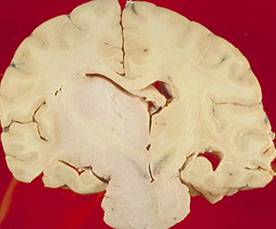
Diffuse Midline Glioma
WHO IV; mostly in kiddos
- even if not a grade IV, will behave badly
Gross: expands pons and basilar artery
Micro: diffusely infiltrating astrocytoma
Genes: H3 K27M mutations
Px: poor
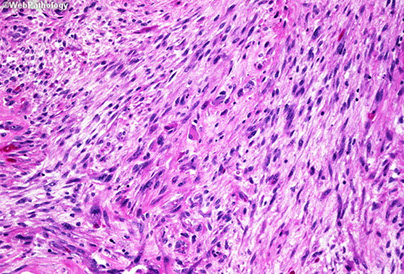
Glioblastoma with Sarcomatous Elements
- aka Gliosarcoma
WHO IV; is a biphasic tumor c glial and mesenchymal components
- 2% of GBM; older pts, 2M>1F
- variant of GBM
IHC: GFAP + in glial part
- malignant mesenchyme is GFAP- neg, is perivascular and has lots of reticulin fibers
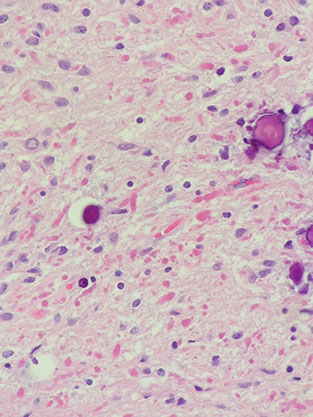
Pilocytic Astrocytoma


PA c EGBs
SEGA
Angiocentric Glioma

Granular cell astrocytoma
Pilomyxoid astrocytoma

Infiltrating Astrocytoma
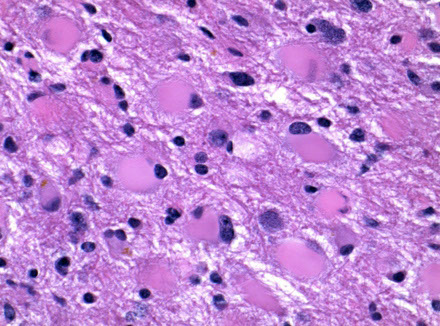
Pleomorphic Xanthoastrocytoma (PXA)


Glioblastoma
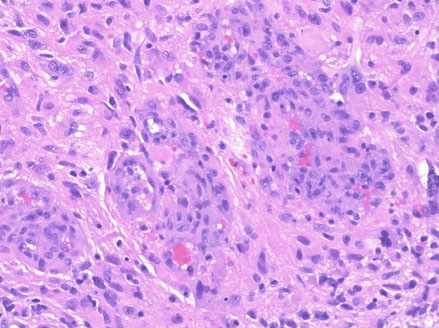
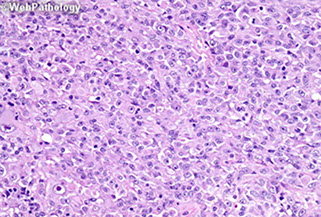
Epithelioid GBM - BRAF V600E

Diffuse Midline Glioma
Diffuse Midline Glioma

Gliosarcoma
Oligodendrocytic Glial Tumors
Oligodendroglioma
MC in 30's in the cerebral hemispheres, present c seizures, usually superficial in the cerebral hemisphere (cortex)
- MUST have 1p/19q co-deletion to dx as oligodendroglioma
- WHO II, unless has inc mits, or look too anaplastic
Imaging shows a discrete mass that does not enhance with contrast admin and is often cortical-based
Micro: Infiltrating glioma with nuclei that resemble oligodendroglia (round, mildly atypical nuclei, not bean-shaped)
- can see clear perinuclear halos ("fried-egg", artifact of formalin fixation [not seen on RFS])
- background of delicate "chicken-wire" capillary network and dystrophic calcifications
The neoplastic cells cluster around neurons, dubbed "perineuronal satellitosis"
- can have areas that look like oligodendroglioma
- tumor cells also seem to cluster (pretty unique characteristic)
May see occasional mits, but are not markedly increased
- variably expresses GFAP
Genetics: Co-deletion of 1p and 19q through FISH or PCR on paraffin-embedded tissue
- have a better response to chemo if this deletion is present
- IDH mutations ??
Px: favorable c 1p/19q loss, Ki67 <5%, and younger pt age
Anaplastic oligodendroglioma
WHO III, Has similar features as oligodendrogliomas, but with increased cellularity, increased atypia, microvasc prolif, small foci of necrosis and frequent mits (>6 /hpf)
Genes: should have 1p/19q co-deletion, if not will have ATRX mutation and be indicative of an astrocytic tumor
ATRX (alpha-thalassemia/mental retardation syndrome X-linked)
- ATRX and DAXX involved in telomerase maintenance, and ATRX mutated in about one third of pediatric and adult gliomas
- ATRX mutation and 1p 19q co-deletion is mutually exclusive
- IDH-1 mutated gliomas with ATRX mutation have good px
Mixed gliomas
Waste-basket diagnostic category
Shows a mix of both astrocytic (bean-shaped nuclei) and oligodendroglial (round nuceli)
- is sometimes called an oligoastrocytoma
In the presence of mostly astrocytes, a 1p/19q LOH is uncommon
Oligodendroglioma
Anaplastic oligodendroglioma

Subependymoma
Subependymoma
Low grade (WHO grade 1) tumor often found incidentally at autopsy; MC in the medulla of 4th ventricle
Micro: see exophytic intraventricular masses and loose clumps of round monotonous nuclei embedded in a finely-fibrillar, lightly eosinophilic background
- microcysts common
Although ependymal, true or pseudorossettes are rare or absent
- dx rests on recognizing the pattern of clumped, rounded nuclei in a fibrillar background
- since the tumor does not infiltrate, no neurons or axons become entrapped
- up to 1/5 subependymomas have some component of classic or anaplastic ependymoma, but only change the diagnosis to one of these if they make up more than 10% of the total tumor
IHC: (+) GFAP
Myxopapillary Ependymoma
Conus medullaris / cauda equina mass with unique features, WHO grade 1; originate from filum terminale
Micro: No true rosettes or perivascular pseudo rosettes
- bland, monotonous round to spindled cells lining pseudopapillae with hyalinized vascular cores and pools of lightly basophilic, Alcian blue positive mucin
- mits rare/absent and little atypia or signs of invasion
IHC: (+) GFAP, D2-40 (var), S100 (var)
- neg: CK, EMA, Brachyury
DDx: other tumor of filum terminale: nerve sheath tumors, meningiomas, paragangliomas
Tx: can be easily excised
Ependymoma
- the classical ependymoma, which is relatively common and affects mostly kiddos but also adults, usually in posterior fossa (can fill 4th ventricle, causing hydrocephalus)
- slow-growing and originating in the native ependymal linings (ventricles and spinal cord)
- spinal cord lesions MC in adults
- notorious for "drop metastasis" (should do CDF cytology to r/o CSF pathway dissemination of tumor)
WHO grade 2, but can be 3 with lots of atypical features
On imaging, there is a non-enhancing, non-infiltrative mass next to or within ventricles or spinal cord
Micro: variably cellular, non-infiltrative glioma with rounded non-descript nuclei, salt'n peppa chromatin
- characteristically see perivascular pseudorosettes (ependymal cells projecting their cytoplasmic processes towards a central capillary, resulting in a perinuclear, fibrillar anuclear zone) and, less often, true ependymal rosettes which are centrally oriented cellular processes with a true central lumen
- may see ependymal rosette
- a sharp brain-tumor interface is also a characteristic feature
IHC: (+) GFAP [diffuse, processes my radiate to BVs], dot-like perinuclear EMA reactivity highlighting intracytoplasmic lumina / canaliculi (a finding on EM)
- negative NFP
Genes: L1CAM (which can be seen in other tumors) correlates with RELA fusion, a poor px indicator in supratentorial ependymomas
Px: good in adults after complete resection; tends to recur in kids
Clear cell ependymoma
An ependymoma that looks like oligodendroglioma
Seen in kids and young adults supratentorially, although not typically next to ventricle
Micro: round cells with perinuclear clearing (very similar to oligodendroglioma)
- differentiate from oligodendroglioma bc clear cell ependymomas are not infiltrative and also lack the 1p/19q co-deletion
C11orf95-RELA Fusion Ependymoma
- type of clear cell ependymoma that has a worse px
Tanycytic ependymoma can look spindly
Anaplastic ependymoma
Grade III; No significant differences in age or location from WHO II ependymoma
Micro: no defined, objective (?) criteria for this diagnosis
- will see increased cellularity, inc nuclear atypia, frequent mits, coagulative necrosis, inc Ki67
- can still see dot-like perinuclear EMA, diffuse GFAP
Small foci of hypercellularity and inc atypia can be seen in WHO grate II ependymomas and do not qualify for anaplasia
- still not infiltrative
Now there is molecular subtyping of ependymomas:
Supratentorial - Rela-fusion ependymoma bc has fusion bwt RELA gene and C11orf95
Posterior fossa - p65
Spinal - NFK pathway activation
Myxopapillary Ependymoma

Ependymoma - Perivascular pseudorosettes
Ependymoma - perinuc dot-like EMA
Ependymoma - GFAP, ependymal cells projecting cytoplasmic processes into a BV
Clear Cell Ependymoma
Anaplastic Ependymoma

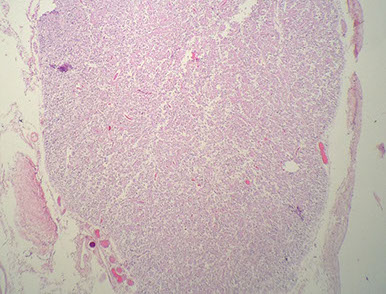
Choroid Plexus Papilloma
WHO grade I
Tumor closely resembles normal choroid plexus, except it forms a mass
- can see hydrocephalus due to CSF overproduction or obstruction, although generally behaves in a benign manner
85% seen in kids (~10 yo), intraventricular (lateral ventricles) tumor
- can occur in the 4th ventricle of adults
Micro: Benign papillary tumor
- distinguish from normal choroid plexus by closely inspecting the lining epithelium
-- normal choroid plexus is lined by taller columnar epithlium which is smooth, but lacks the undulating surface of normal choroid plexus epithelium which is smooth and lacks the undulating surface of normal choroid plexus
- there should be no signoficant atypia and mits should be absent or rare
DDx: carcinoma mets, myxopapillary ependymoma, papillary ependymoma (>1 cell layer), papillary meningioma (no mucin, solid c foci of whorled cells)
IHC: (+) CAM5.2 (94%), transthyretin (89%), GFAP, Kir71
- usually negative p53 and CK20
- CAM5.2 good bc ventricle only epithelial structure in brain, which can stain c CAM5.2
Atypical Choroid Plexus Papilloma
WHO grade II
Intermediate between benign and malignant
Clinically seen in younger pts (10-12 yo) and is intraventricular
Has inc nuclear atypia and easily identifiable mits
Choroid plexus carcinoma
Malignant tumor MC in infants (2-10 yo)
WHO grade III
Micro: malig tumor of choroid plexus derivation
Papillary architecture may be difficult to appreciate with solid sheets of malignant appearing cells and focal papillae
- freq atypical Mits
May see brain invasion and necrosis
Tumors may be congenital.
Imaging: Imaging: mass in ventricle, appears to invade into brain
- should have Flair around lesion, which is indicative of invasion
IHC: (+) Cam5.2 and GFAP
*** all other tumors should be cam5.2 negative... this bridges the gap
Px: very poor
Ganglioglioma
Has both dysplastic neurons (ganglion cells) and atypical glial components
- need ganglion component and glial component
- the "glial" component is usually astrocytoma, rarely oligodendroglioma
MC in temporal lobes and is assoc c seizures and are rare in kids and adults
Imaging: cystic superficial mass with occasionally with enhancing mural nodules
Micro:
- Dysplastic neuronal component has "ganglion cells" (neurons) that are abnormally clustered, pleomorphic and occasionally binucleated. Must exclude entrapped normal neurons (innocent bystanders) entrapped by an infiltrating glioma
-Atypical glial component: Atypical infiltrating glial component of varying degree of malignancy, graded as you would an astrocytic neoplasm without a ganglionic component
- may see eosinophilic granular bodies (EGBs)and Rosenthal fibers (similar to those seen in pilocytic astrocytoma)
- ganglion cells may be sparse
IHC: (+) SYN, Sm32, NeuN, GFAP
Genes: subset may harbor BRAF V600E
DDx: trapped normal cortical neurons in a diffuse astrocytoma may be mistaken for neoplastic ganglion cells
Tx: resection
Px: if the background glial component looks bad (rare) may behave worse
Desmoplastic infantile ganglioglioma/astrocytoma
Rare tumor that looks like the name implies
Seen in kiddos <2 yo c variable imaging findings; usually very large
Micro: atypical mixed glial and neuronal tumor c prominent desmoplasia which gets highlighted with reticulin
- invade subarachnoid space (similar to JPA, PCA, GG)
Tx: Resection
Pc: good (if young age and small tumor size)
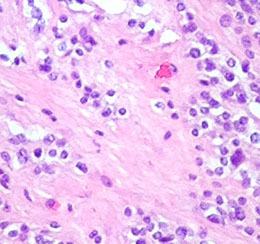
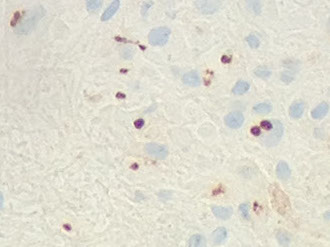
Dysembryoplastic Neuroepithelial Tumor (DNET)
Rare glioneuronal tumor of teens and adolescents (<20 yo; M>F) with hx of intractable seizures
-low grade, intracortical, multinodular
*** What do you do if you see a floating neuron? Get D-Net ***
Imaging: supratentorial nodular mass often in the temporal lobe adjacent to amygdala
- multinodular, intracortical, non-enhancing mass
Micro: notable amount of intratumoral heterogeneity and nodularity
- often see areas indistinguishable from ependymoma, oligodendroglioma, etx. 2 highly suggestive features: 1) Floating neurons (neurons which appear to float in pools of mucin; 2) Specific glioneuronal element (linear arrays of oligodendroglioma-like cells [OLCs]) look like oligos
- can have glial nodules
To dx this tumor you need hx of seizures, location, nodularity and histologic findings
- by some this tumor is considered a malformation (not neoplastic), esp in the light that some are assoc c cortical dysplasia
IHC: (+) S100, NeuN (focal)
- neg: SYN
Px: B9, excellent px even if not completely taken out
Choroid Plexus Papilloma


Atypical choroid plexus papilloma

Choroid plexus carcinoma
Ganglioglioma

Dysembryoplastic Neuroepithelial Tumor (DNT) - floating neurons
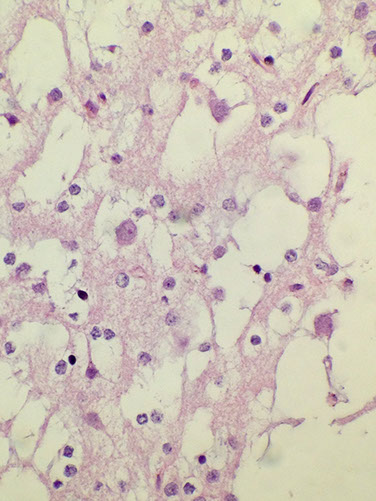
Rare Neurocytic Tumors
Central Neurocytoma
MC in teens and young adults (~30 yo); comes from intraventricular septum, mimic oligodendrogliomas
Imaging: Intraventricular mass adherent to septum pellucidum or near foramen of Monro
Micro: Composed of "neurocytic" cells with round, monotonous nuclei with finely stippled salt and pepper-like chromatin
- often have a perinuclear halo and look very similar to oligodendroglioma; islands of slightly fibrillar, eosinophilic neuropil amongst the tumor cells may help
- can see frank ganglionic differentiation
- 2 keys to dx: strong diffuse synaptophysin and neu-N staining and 2) intraventricular location
-- does not infiltrate brain parenchyma
All previously diagnosed "intraventricular oligodendrogliomas" in literature are most likely central neurocytomas
IHC: (+) SYN (diffusely +), NeuN (var)
- neg: GFAP (sometimes pos)
Px: good, but recurs if residual tumor left after resection
Gangliocytoma
Rare tumor in kids and young adults that present with seizures
Imaging: tumor most often seen in temporal lobe
Micro: Non-infiltrative tumore consisting solely of dysplastic ganglion cells (pleomorphic, abnormally clustered, and binucleated)
- lacks malignant features
- think of it as a ganglioglioma without the atypical glial component
IHC: (+) SYN, SM32, NeuN
Dysplastic Gangliocytoma of the Cerebellum
- aka Lhermitte-Duclos disease
WHO grade I
Rare lesion that unilaterally enlarges the cerebellum; hypertrophy of internal granule cells c assoc secondary myelination of molecular layer
- assoc c Cowden syndrome (10q PTEN gene mutation)
- thought to be a hamartomatous growth of the cerebellum
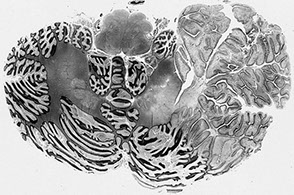
Imaging: tiger stripes on MRI
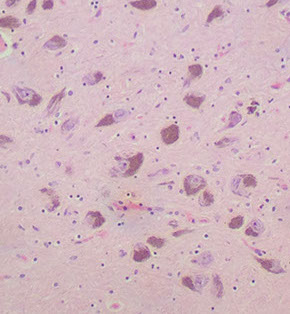
Histo: replaces the internal granular layer of the cerebellar folia c moderate to large hypertrophic ganglion cells
- also look for decreased number of Purkinje cells, microcalcifications, large bizarre neurons, capillary networks, and vacuolization of the cerebellar white matter
IHC: neurons highlighted c neurofilament, chromogranin, and synaptophysin
- tumor cells negative for GFAP (though may see an astrocytosis highlighted)
Genes: Cowden syndrome assoc c PTEN mutations
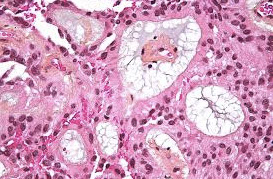
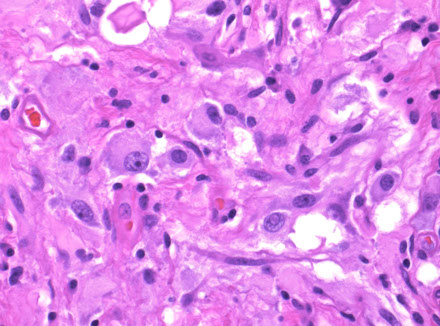
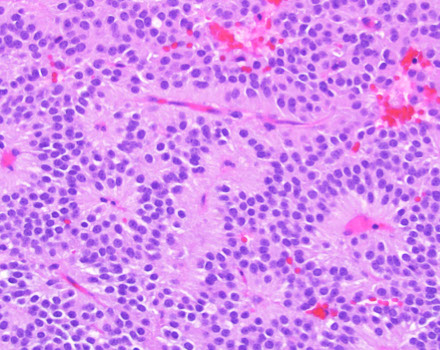
Central neurocytoma
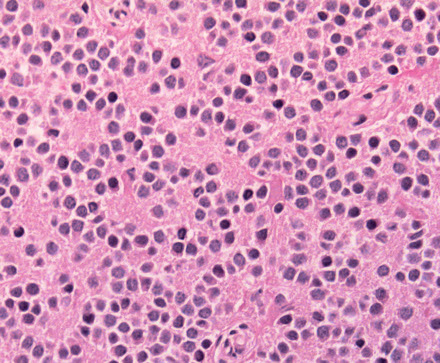
Composite gangliocytoma (black arrow) and sparsely granulated somatotroph adenoma (outline arrow) are shown. Paranuclear fibrous bodies (white arrow) seen in adenoma cells.
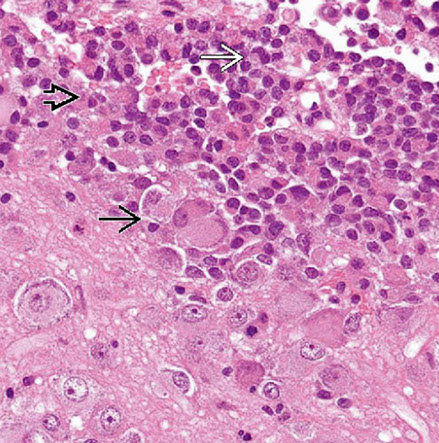
Lhermitte-Duclos
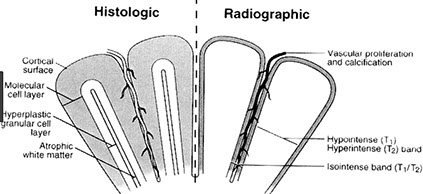
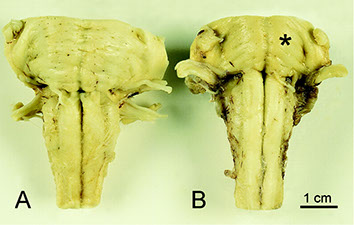
Pineal Region Tumors - Perinaud's Syndrome
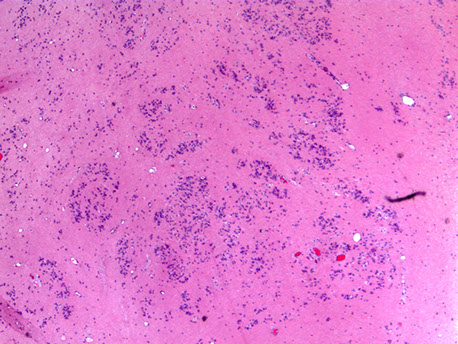
Germ cell tumor
- aka pineal germinoma
MCC tumor in pineal region; M>F; usually germinoma; Look like germ cell tumors of the testes
- can be assoc c granuloma formation
Parinaud Syndrome: dorsal midbrain (pineal region) lesion
- vertical gaze palsy, convergence retraction nystagmus, and light-near dissocation
IHC: (+) OCT3/4 PLAP, c-KIT CD117)
Can be other germ cell tumors - Embryonal carcinoma, yolk sac tumor, choriocarcinoma, teratoma
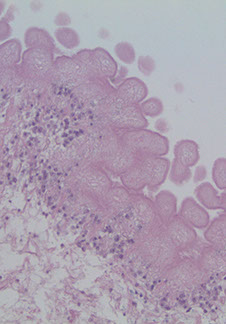
Pineocytoma
Rare tumor MC in adults
Imaging: Homogenously contrast enhancing; can be solid or cystic
Micro: sheets of small, monotonous cells that look like normal pineocytes
- large "pineocytomatous" rosettes are often seen; no/few mits; can see degenerative atypia in benign tumors
- synaptophysin and neurofilament (+)
Pineal Parenchymal Tumor (Neoplasm) of Intermediate Differentiation (PPTID)
Intermediate bwt pineocytoma and pineoblastoma
- higher cllularity and atypia and mits and Ki67, but no real general consensus yet
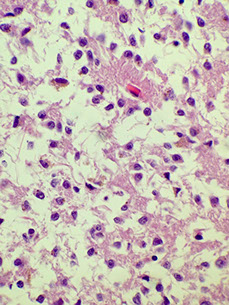
Pineoblastoma
Very malignant beast, seen in kiddos, spreads through CSF
- WHO IV
In the most primitive end of pineal neoplastic spectrum, comprising a small-round blue cell tumor with small rosettes (not the large "pineocytomatous" of benign tumors)
Lots of mits, inc Ki67; (+) synapto and NFP
Germ cell tumor pineal gland


Pineal Germ Cell tumor
Pineocytoma
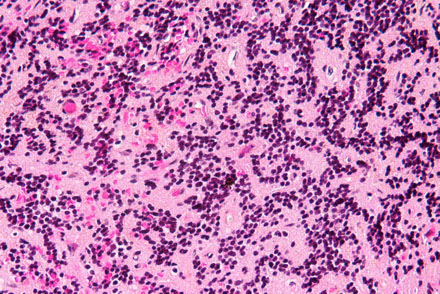
Pineoblastoma
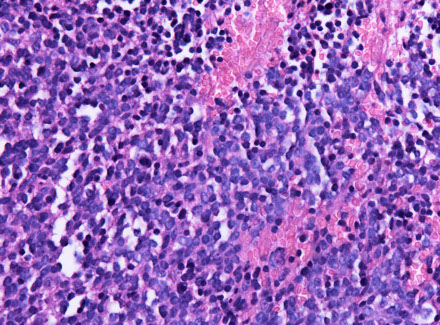
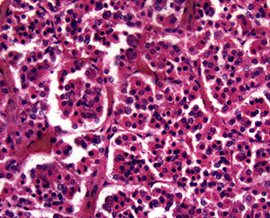
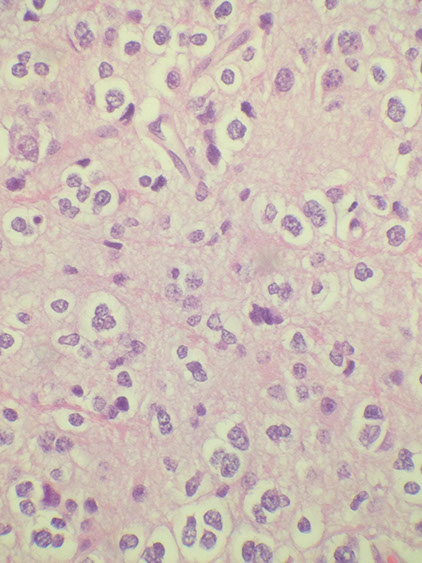
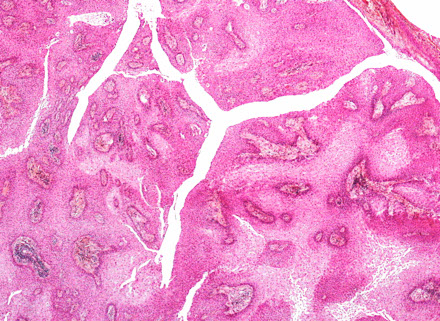
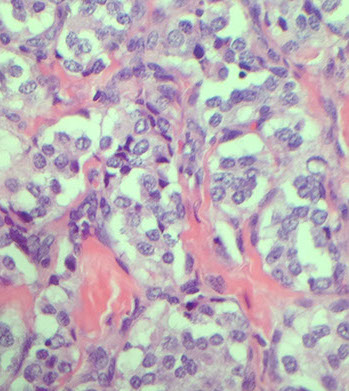
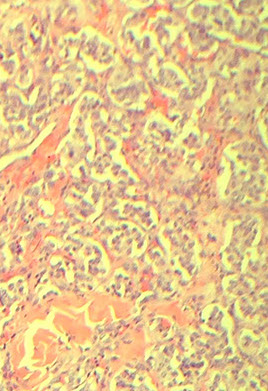
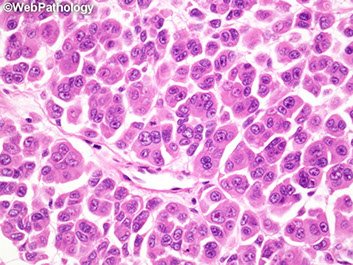
Pituitary Adenoma
MC sellar region tumor; found in young adults and elderly; usually B9, malig changes rare
- < 1 cm = microadenoma; >1 cm = macroadenoma
Macroadenomas don't secrete, so they are silent until they produce sx such as headache, bitemporal hemianopsia
- may be part of MEN type I, and can have G-protein mutations (seen in thyroid and parathyroid adenomas)
See monotonous prolif of round epithelioid cells c variable nuclear atypia. Key features:
- sheet-like growth pattern
- loss of normal small-nest architecture
- monotonous cells
- expansion/effacement of the normal reticulin network of the adenohypophysis (seen on reticulin stain)
If there is lots of bleeding into an adenoma, known as pituitary apoplexy
- calcifications and deposition of amyloid-like substance can be seen (prolactinoma)
Lactotroph (prl cell) adenoma
Prolactinoma
- most freq hyperfunctioning adenoma (~1/3)
- can have focal calcifications (psammoma bodies) and amyloid
- may cause infertility, +/- galactorrhea
- Forbes-Albright syndrome: galactorrhea and amenorrhea in pt c prolactin-secreting adenoma
- Outside of pregnancy, inc prolictin is most commonly the result of a pituitary tumor. In women, this causes amenorrhea and/or galactorrhea.
- In men, the symptoms are much less dramatic and loss of libido is the only common symptom (but may be absent in many cases).
- Increased prolactin may also be due to compression of the pituitary stalk or hypothalamic damage, due to loss of inhibition by dopamine, the major prolactin inhibitory factor. Drugs that decrease dopamine production or action (phenothiazines, H2 antagonists and antidepressants) also increase prolactin. Prolactin levels > 200 ng/ml (> 10x normal) almost always indicate prolactinproducing tumors, while levels between normal and 100 ng/mL are often due to drugs or pituitary stalk compression.
Somatotrophic (GH cell) adenoma
- produces excess GH
- 2nd MCC functioning pituitary adenoma
- 2/5 caused by mutation GTP-ase deficient a-subunit of G-protein
- can cause gigantism or acromegaly
~1/3 of GH-producing adenomas are bihormonal and also secrete PRL
- GH overproduction virtually always the result of a pituitary tumor. Children who produce excess GH develop gigantism, while adults become acromegalic. Excess GH also causes hyperglycemia in about 50% of cases. Bc GH levels fluctuate and may be only mildly elevated, a better test for GH overproduction is insulin-like growth factor I (IGF-I, somatomedin) and/or IGF binding proteins.
- GH stimulates the liver to manufacture IGF-I and its binding proteins; because of their long half-life, a single determination is usually all that is necessary. Some prefer to measure GH after administering glucose (glucola), since hyperglycemia inhibits normal growth hormone production; this is less sensitive than IGF-I measurement. About 75% of tumors show paradoxical increase in GH after TRH administration.
Corticotroph (ACTH/MSH cell) Adenoma
- usually a microadenoma in the median wedge that may cause Cushing disease (excess ACTH cayses bilateral adrenal cortical hyperplasia c hypercortisolism
- Crooke's hyaline change - lots of cytokeratin in extratumoral pituitary cells (can be seen in all causes of hypercortisolism)
- Nelson's syndrome - rapidly enlarging pituitary tumor c skin hyperpigmentation in Cushing dz pts treated previously c bilateral adrenalectomy
Null cell adenoma
- 1/4 of pituitary adenomas, aka chromophobe adenoma
- IHC are negative, are the largest pituitary tumors, usually of gonadotrophic lineage
- Gonadotropin overproduction seen in about 20- 25% of pituitary tumors. Many lesions formerly thought to be nonfunctional produce either intact gonadotropins or their subunits. Like TSH and HCG, FSH and LH have a common a subunit and distinct b subunits; the intact hormone is active. Overproduction of gonadotropins usually causes a clinical picture of hypogonadism, since steady release of gonadotropins inhibits normal function.
- Tumors often show paradoxical secretion in response to TRH.
IHC: diffuse, strong CAM5.2 and synaptophysin; may also see (+) stain depending on specific hormone secreted (GH/LH etc)
"Stalk effect": a large pituitary can press on the pituitary stalk and interrupt the normal dopamine flow to the adenohypophysis, resulting in mild elevations of prolactin. Thus, a low-level of serum prolactin does not necessarily signify a prolactinoma
Craniopharyngioma
Rare, Grade I, in sellar region of kiddos, that is slow- growing and can damage hypothalamus and compress optic chiasm, usually near the 3rd ventricle which can impede CSF cycle causing inc pressure in different areas of brain (hydrocephalus)
- derived from epithelial remnants of Rathke pouch
- usually suprasellar
Gross: partly solid (c calcifications and "machine-oil" liquid) and partly cystic components
Histo:
2 distinct microscopic patterns and ages of presentation:
1) Adamantinomatous: tumor of young adults, sellar region, cystic, contains "motor oil" 2/2 proteins in blood and cholesterol crystals)
- ameloblastic-like epithelium (peripheral palisading [squamous nests surrounded by columnar cells], stellate reticulum [loose squamous nests])
- wet keratinization (clumps of anuclear, "ghost" keratinocytes
- dystrophic calcification on CT
- finger-like extension into adjacent brain parenchyma
Genes: mutations in CTNNB1 encoding B-catenin (can be tested for using IHC, which shows accumulation of stain in nuclei)
2) Papillary craniopharyngioma: older pts, more posterior (in 3rd ventricle), non-cystic
- bland squamous prolif c focally papillary architecture (resembles squamous papilloma seen elsewhere in the body)
- no keratinization
- no calcification
- easier to resect and recur less often
- Genes: mutations in BRAF V600E
IHC:
- negative CK8/20
Other causes of sellar-region masses:
Langerhan's cell histiocytosis (S100+, CD1a+, lots of eos)
Germinoma: can be paucicellular and granulomatous
Metastatic carcinoma
Lymphocytic hypophysitis: inflam tumor of sella; dx of exclusion
Neurohypophyseal tumors (pituicytoma, granular cell tumor)
Pituitary Adenoma
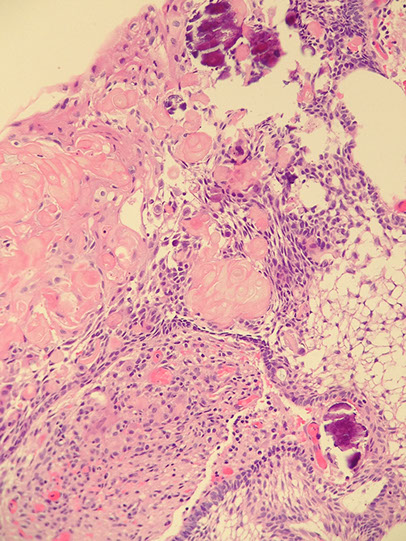
Adamatinomatous Craniopharyngioma
wet ------>
keratin
<--calc
stellate reticulum
Papillary Craniopharyngioma
Pallisading -> cells
Medulloblastoma / Primitive Neuroectodermal Tumor (PNET)
Malignant, common, found in posterior fossa in kiddos (7 yo), esp on vermis (cerebellar midline) or 4th ventricle where it grows rapidly and can cause obstruction and hydrocephalus
- made of primitive neuroepithelial cells in embryonic and fetal nervous system
- very rare in adults
- notorious for "drop metastasis" (should do CSF cytology to r/o CSF pathway dissemination of tumor) - also seen c ependymoma
Micro: small round blue cells c high mits and pyknosis and karyorrhexis
- can see Homer-Wright rosettes (neuropil in center, no true vessel lumen)
- several variants: desmoplastic/nodular; anaplastic; large cell; medullomyeloblastoma; melanotic medulloblastoma
Desmoplastic / nodular medulloblastoma occurs in older pts (~18 yo vs 8 yo for classic variant) and is assoc c nevoid basal cell carcinoma syndrome (Gorlin syndrome)
- IHC: reticulin poor "pale islands" expessing SYN
- Genes: PTCH gene (cr 22) mutations (hedghog pathway); better px
- Type 2 Turcot Syndrome: FAP + medulloblastoma (APC gene, 5q21)
- may have better px bc able to mature in reticulin nodules
- Nodules and intranodular desmoplasia assoc c SHH-activation
Large cell / Anaplastic type - has cell wrapping
- worse px
IHC: (+) GFAP, SYN (both are [+] 2/2 overlap bwt neuronal and glial differentiation), CD99 (membranous)
- neg neuronal
Genes: N-MYC mutations assoc c some subtypes of medulloblastoma, assoc c poor px
t!11;22) EWSR1-FLI1
- EWSR1-ERG
NOTE: a soft tissue PNET/Ewing's sarcoma and CNS PNET are not related!!! (different genetics and everything else!!!)
Px: Better if localized to cerebellum, pt > 3 yo, no drop mets
- Worse if drop mets, brain stem invasion, and MYC gene amplification
- also poor px in younger pts bc cannot tx c radiation (considered high risk) and usually get drop mets
Atypical Teratoid Rhabdoid Tumor (AT/RT)
Rare, extremely aggressive; found in young kiddos (about 1-2 yo)
- arises in cerebellum (can mimic medulloblastom) or cerebrum
- rhabdoid morphology not always appreciable; may be spindly or myxoid
- mets throught the CSF
Can be both infra and supra-tentorial (about equal incidence)
- if infra- will be clinically similar to medulloblastoma
Highly atypical cells with bad nuclear features
- has rhabdoid morphology: eccentric eosinophilic cytoplasm distended by homogenous eosinophilic cytoplasmic inclusion that pushes into nucleus
- usually apoptotic bodies present, showing that it is high grade
IHC can be highly variable; the one thing that is certain is the characteristic loss of INI1 gene on 22q11.2, which is responsible for maintaining and remodeling the chromatin structure (from SMARCB1) (INI1 = SMARCB1 = BAF47)
--- caveat!- small % of choroid plexus tumors can have loss of INI1
- BAF47 stains cells with a working INI1; so negative BAF47 is characteristic of AT/RT (same for hSNF5?)
- EMA positivity may help to differentiate from PNET
- BRG1 (still being researched...) may be more specific
"Teratoid" bc stains like teratoma (can be GFAP, SMA, EMA) showing different lineages
Rhabdoid Tumor Progression Syndrome (RTPS) -
Medulloepithelioma
who grade IV
Small round blue cell tumor usually in kiddos bwt 6 mo and 5 yrs, M=F, occurs in cerebral hemispheres, brainstem, cerebellum
- can resemble primitive neutroal tube or photoreceptors, forming Flexner-Wintersteiner rosettes
Embryonal Tumors with Multilayer Rosettes (ETMR)
- previously known as embryonal tumor with abundant neuropil and true rosetts (ETANTR); and also encompasses CNS PNET
Rare small round cell tumors; in kiddos <4 yo, mostly in kiddos <2 yo and is MC in girls; is one of the most aggressive tumors
Imaging: large demarcated solid mass c patchy or no contrast enhancement c surrounding edema usually c significant mass effect; T1 has dec intensity; T2 inc intensity; T1 C+ (Gd) patchy or no contrast enhancement
Micro: undifferentiated neuroepithelial cells resembling those of classic CNS PNET in multilayer rosettes, abundant well-differentiated neuropil and ependymoblastic rosettes scattered throughout paucicellular regions of neoplastic neuropil
- unlike medulloblastoma, ETMR has no epithelial-like formation, but instead has a characteristic ependymoblastic rosettes in both highly cellular as well as acellular areas
IHC: LIN28A overexpressed in at least a subset of tumor cells
Genes: Amplification of the C19MC region of cr 19q13.42
Tx: surgery, systemic chemo, craniospinal rads
Px: dismal; most die within 9 mo
DDx: medulloblastoma; medulloepithelioma; AT/RT

Medulloblastoma / PNET

Medulloblastoma with Homer-Wright rosette

Desmoplastic/ nodular medulloblastoma

AT/RT

ETMR

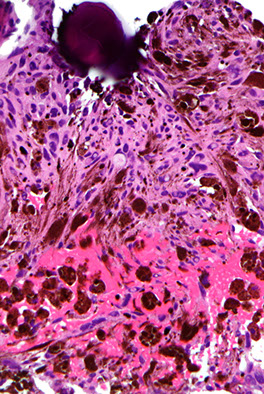
Chordoma
Low-grade yet aggressive tumor of notochordal remnants
An expansile, destructive lesion MC in clivus (base of skull) and sacral / coccygeal regions
Micro: Epithelioid neoplasm c cords of "physaliferous cells" c vacuolated cytoplasm -- "physaliferous" = "bubble-bearing"
- myxoid stroma background
- can have well-developed chondroid features
- infiltrates bone
Must distinguish from low-grade chondrosarcoma c cytokeratin (Chordoma is (+))
IHC: (+) CKs, EMA, S100, Brachyury
- neg: GFAP, D2-40
Cells of intraosseous benign notochordal cell tumor have the same IHC as chordoma, although in that tumor there is a lack of lobular architecture, fibrous bands and myxoid matrix
DDx: ecchordosis physaliphora (looks like a chordoma, but does not invade or destruct)
- chondrosarcoma also seen in base of brain
Chondrosarcoma
Low-grades can look like a chordoma and occur in similar places, but will be CK -
Giant Notochordal Hamartoma
Intra-osseus lesion of the clivus
Developmental in origin
Non-aggressive radiologic features
Micro: bland chords of physaliferous cells amongst trabeculae of bone
Ecchordosis Physaliphora
Presents as nodule of firm tissue adherent to basal artery
Micro: indistinguishable form chordoma
- only way to distinguish is from imaging
Chordoma
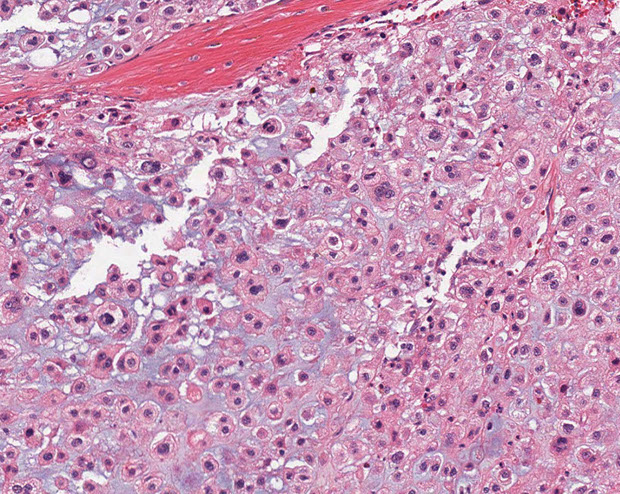
Chordoma - with physaliferous (bubble-bearing) cells
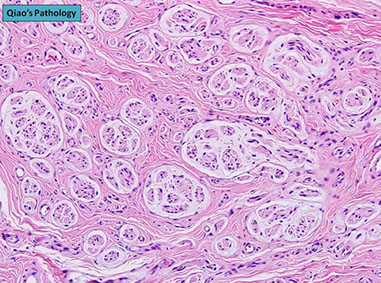
Meningioma
Generally, meningiomas arise from "meningothelial (arachnoid) rests" - small nodules of meningothelial tissue that are present throughout the leptomeninges, pachymeninges and the choroid plexus and cerebellopontine angle (2nd MCC cpa tumors [3%])
- most are grade I
-- can classify based on behavior (brain invasion assoc c higher grade [at least grade II], whereas invasion in bone and scalp can be seen in grade I)
*** CCPR = the bad meningiomas ***
*** Clear cell, Chordoid, Papillary, Rhabdoid ***
Though many subtypes exist, similarities exist:
- intranuclear pseudoinclusions (not exclusive to meningioma, but are the most helpful feature, esp during RFS's)
- intralesional collagen deposition (not many primary CNS tumors make collagen; but the MC are meningiomas)
- localization (consider meningioma first in a dural-based mass)
- F>M, at about 9:1 (CNS tumors are usually M>F); older pts
- common feature is whorls and psammoma bodies
No good specific IHCs, the best is probably Epithelial Membrane Antigen (EMA), which is patchy and unreliable; may have inc EMA as it gets older, but strong vimentin (+), PR (fairly specific, + in 2/3) and S100 (+)
- nearly all are positive for somatostatin receptor 2a (SSTR2A)
- negative GFAP, OCT4
Genetics: monosomy cr 22 (thus assoc c NF2)
DDx: dural-based lesions (can be hemangiopericytoma)
Meningothelial Meningioma
MC subtype
Has meningothelial whorls and psammoma bodies (the calcified corpses of meningothelial whorls); should not see atypical features
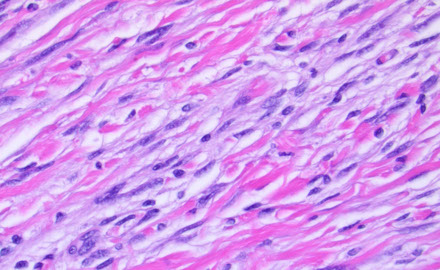
Fibrous meningioma
Collagen-rich, spindled tumor
Whorls and psammoma bodies help to dx, but are not req'd
IHC: (+) S100 (4/5), EMA, PR
- neg: CD34, bcl-2, Melan A
Psammomatous Meningioma
A type of meningothelial meningioma where psammoma bodies predominate
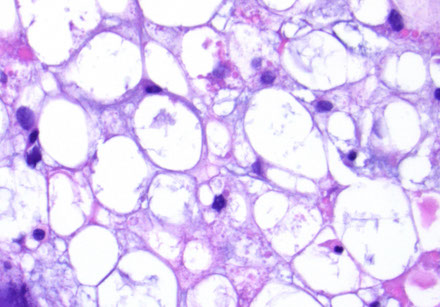
Secretory meningioma
Subtype with "pseudopsammoma bodies", which are eosinophilic round concretions and not the usually blue ones
- these stain (+) c PAS and CEA and are also focally keratin (+)
IHC: (+) CK (1/2+), CEA and PAS pos in cytoplasmic inclusions
Genes: KLF4 and TRAF7
Meningothelial Meningioma


Fibrous meningioma

Secretory Meningioma
Atypical meningioma
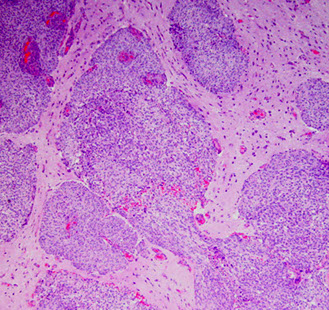
More aggressive tumor due to presence of brain invasion (most important criteria in dx'ing), inc mits (from 4 up to 20 per 10 hpfs), prominent nucleoli (macronucleoli), sheet-like growth pattern, inc NC ratio, inc cellularity, spontaneous necrosis
- it is ok if it invades the skull, but not the brain itself
- WHO grade II

Atypical meningioma - brain invasion
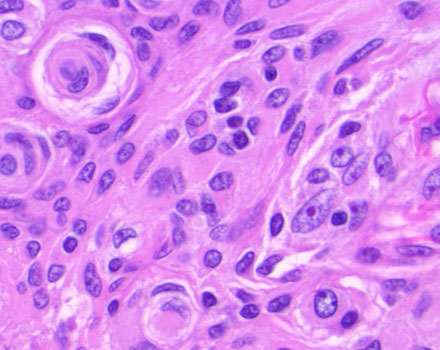
Clear cell Meningioma
Micro: round cells c clear cytoplasm amongst thick ropey bundles of eosinophilic collagen
- extracellular tyrosine crystals have been seen in this tumor
- often lacks the characteristic features of meningothelial differentiation
- must ddx from clear cell renal cell carcinoma
- WHO grade II
Clear Cell Meningioma
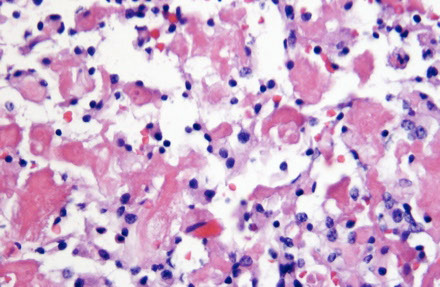
Chordoid meningioma
Dural lesion; usually underneath 3rd ventricle
Micro: nests and chords of epithelioid cells c cytoplasmic vacuolization (physaliferous cells - resembling chordoma of clivus/sacrum) in a myxoid background
- presence of focal meningothelial featyres (whorls, Psammoma bodies) confirms meningothelial component
- WHO grade II
IHC: (+) EMA, D2-40
(-) S-100 and CK, and GFAP, Brachyury
Chordoid Meningioma
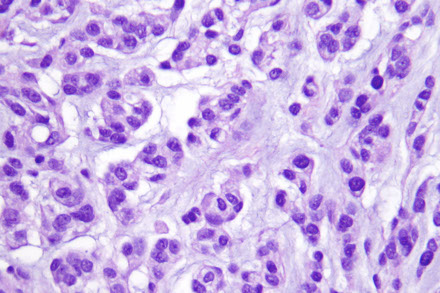
Papillary meningioma
A more aggressive variant MC in children
- WHO grade III
Micro: cellular discohesion resulting in a papillary appearance in which atypical menigothelial cells surround vascular cores
- NOT a true pappilary lesion, just has a little pappilary feature in it
- lots o mits
Papillary meningioma
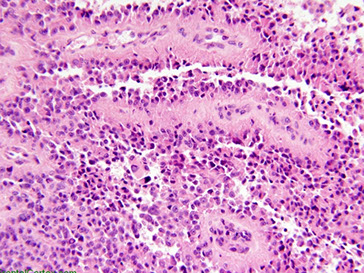
Rhabdoid Meningioma
More aggressive
- WHO grade III
Micro: Majority of tumor with cytologic atypia, hypercellularity, and tumor cells c rhabdoid features (eccentric nucleus, red cytoplasmic inclusions that push on the nucleus, with red, fibrillar cytoplasmic inclusions)
small amounts of rhabdoid features are not diagnostic of this entity but should raise worries
Rhabdoid meningioma
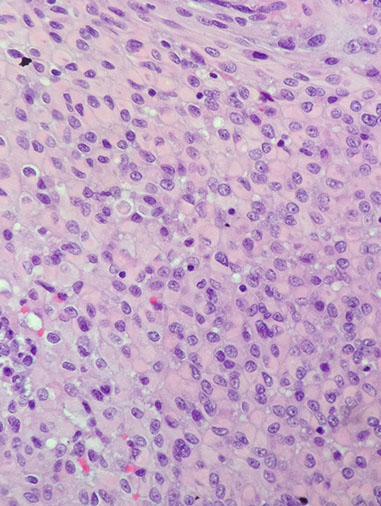
Anaplastic meningioma
20+ mits /hps
Anaplastic cytology: all the bad features
- WHO grade III
Anaplastic criteria
Hemangiopericytoma
May be mistaken for meningioma due to similar clinical and radiologic finding, and are thus treated c embolization followed by surgery
- tend to occur at younger ages than meningioma
- should suspect this in a spinal lesion in a younger male, even before frozen section
same as HPC in other parts of body, with characteristic staghorn vessels, abundant cellularity with ovoid appearance
- patternless pattern
Call it anaplastic if >5 mits/10 hpf or necrosis or bad nuclear atypia
IHC: Reticulin shows pericellular reticulin network; most have patchy CD34; and a caveat is that it can be EMA (+) --> just like meningiomas, (+) CD99
Px: may behave more poorly in CNS than elsewhere

Hemangiopericytoma
Meningioangiomatosis
Rare, b9 hamartomatous lesion lesion c leptomeningial and meningovascular prolif (can have nuclear inclusions like meningioma)
- usually in 2nd decade
- 1/4 assoc c NF2
Micro: lots of perivasc meningothelial cell prolif c collagen in cortex and sometimes white matter
- also see inc cortical vascularity, leptomeningial calc, variable psammoma bodies and tangles
- no high grade features present
IHC: (+) EMA, vimentin, focal S100 (1/5), variable CD34
Meningioangiomatosis


Rathke's Cleft Cyst
Sellar region (pituitary) cyst lined by respiratory-type epithelium
- 2/2 Rathke's pouch not developing properly; may be found in up to 25% of people!
- change in pressure can cause headaches
Tx: drainage and bx
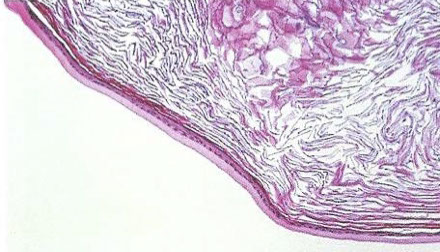
Epidermoid cyst
MC in cerebellopontine angle; mature and ordered sqamous epithelium c pilosebaceous structures
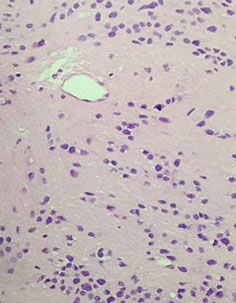
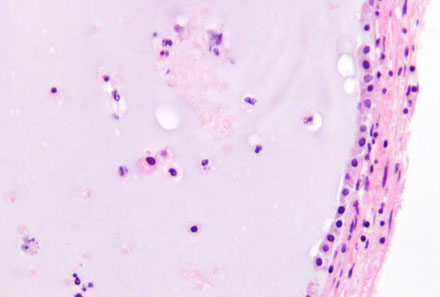
Colloid cyst
Respiratory-type epithelium c central homogenous eosinophilic contents; almost always posterior to foramen of Monro
- can cause obstructive hydrocephalus (from ball-valve effect esp when pt lays flat, thus tend to sleep upright), inc intracranial pressure, headache, vertigo, memory problems, diplopia, brain herniation depends on size, imaging characteristics, size of ventricles, and age of pt
- can also see Rosenthal fibers in cyst wall
Tx: surgery
Enterogenous Cyst
MC in brainstem or spincal cordl development abnormality
Lined by columnar, often mucinous epithelium
- can be ciliated
B9
Arachnoid Cyst
Common, b9 accumulation of CSF under arachnoid layer that comes from reduplication / splitting of meninges
Tx: can do surgery if sx
Cysticercosis
Pork tapeworm (Taenia solium) infx
- forms well-circumscribed cysts in brain, esp in intraventricular region
Can present c seizures, headache, sensory probs, dementia
Calcifications common
3 layers:
Undulating red waxy cuticle; germinal layer; inner loose myxoid layer
Rathke's Cleft Cyst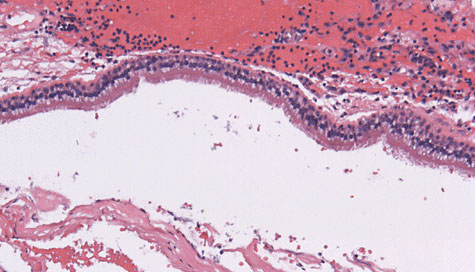
Epidermoid Cyst
Colloid Cyst
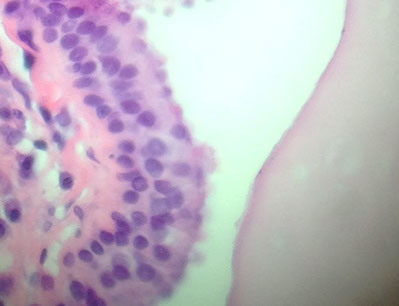

Arachnoid cyst

Cysticercosis
Hemangioblastoma
Adolescents and adults
- Cyst with a mural nodule in the cerebellum of an adult?
= Hemangioblastoma!
- cerebellar hemangioblastoma can secrete erythropoietin (similar to RCC)
Imaging: Cyst c enhancing mural nodule
- often cerebellar, can be cerebellar and retinal
Micro: 2 parts
- Stromal cells in neoplastic part with foamy clear cytoplasm
- delicate capillary network (non-neoplastic?)
~1/4 related to von Hippel Lindau syndrome, due to VHL gene mutation of cr 3p (3 letters in vHL)
- Must be able to distinguish from met clear cell RCC (RCC has +EMA, - inhibin; hemangioblastoma is the opposite)
IHC: (+) nested reticulin positivity, inhibin, Oil red O, Aquaporin-1, NSE
- neg: EMA, CKs, PAX2/8, CAIX, RCC< CD10
DDx: Renal cell carcinoma (should stain negative with RCC markers)
- similar to RCC, hemangioblastoma can secrete erythropoietin
Choroid glioma of the 3rd ventricle
Rare tumor of adults and elderly, MC in females
Imaging: Solid, well-circumscribed contrast-enhancing mass of the 3rd ventricle
Micro: looks just like a chordoma (epitelioid cells lying in chords, mucinous background) BUT occurs in 3rd ventricle and cells strongly GFAP (+) (CK, S100 weak or negative)
Not aggressive, but not able to excise due to location and pts eventually die from hydrocephaly
Must distinguish from Chordoma (S100/CK+, GFAP -) and choroid meningioma (EMA+, GFAP-)
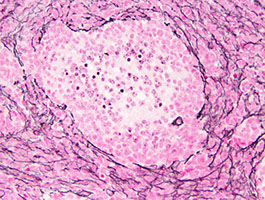
Paraganglioma
Occurs in cauda equina region of spinal cord
- assoc c Carney syndrome, MEN, NF-1, von Hippel-Lindau
Indistinguishable from paragangliomas in other parts
- Head and neck sites: carotid body, jugulotympanic
- Very few head and neck paragangliomas are functional, unlike adrenal tumors.
Carotid body paraganglioma: most common H&N site
- M = F; Wide age range. MC in pts living in high altitudes
- Sx: presents as painless neck mass, large tumors may be assoc c hoarseness, vocal cord paralysis, Horner’s syndrome
- Incidence of malignancy ~ 10% (either local invasion or
mets)
Jugulotympanic paraganglioma: “glomus jugulare tumor;”
- F > M, often in 5th-7th decades. Location and infiltrative nature make recurrence rate around 50%
Px: Malignancy is rare, but they may still be lethal due to
invasion of vital structures.
Has nests (Zellballen)/ trabeculae of epithelioid "chief" cells c "salt and pepper" chromatin and encircling spindled sustentacular cells
- prominent vessels (may confuse c ependymoma)
- no mits (unless high grade) or rosettes
CK+ (vs non-CNS paragangliomas, which are CK-)***
IHC: [same as pheochromocytoma] (+) reticulin highlights nesting pattern, (+) S100 in sustentacular cells, (+) CHR, SYN
- negative CAM 5.2 (can be (+) in cauda equina)
Genes: some have been found to have LOH in up to 1/2
- Succubate dehydrogenase (SDH) mitochondrial complex gene mutations are the MC assoc c b9 paragangliomas of the head and neck
Px: up to 40% malignant and can met
- only way to determine if malignant is if it mets
SpinoCerebellar Ataxia type III (SCA3) / Machado-Joseph disease
Slowly progressive clumsiness, staggering / lurching gate, speech / swallowing difficulty, highly variable presentation
- have DNA repeat (CAG) expansion in ATXN3 gene - affects ataxin-3 protein; dominant inheritance with anticipation
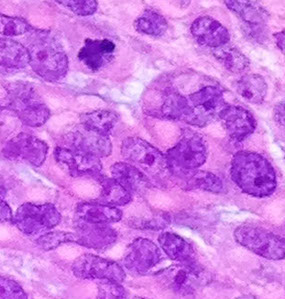
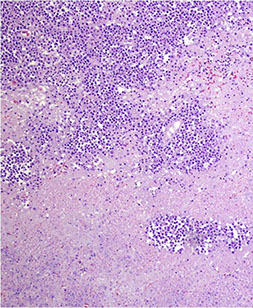
CNS Lymphoma
EBV - associated DLBCL of the CNS
- formerly a Primary CNS lymphoma
- can occur anywhere, but predilection for brain
- active EBV acquired during childhood during infectious mono, with latent phase where EBV persists in B cell nuclei as episomes
- immunosuppression triggers prolif of EBV infected B cells (was seen c AIDS, but not so much now 2/2 HAART tx, but do seen in pts under iatrogenic immunosuppression or in elderly)
Micro: pleomorphic round discohesive sheets. lots o mits and necrosis
- prominent angiocentric and angiodestructive growth, esp at periphery, where they appear to accumulated in perivascular space and are angiodestructive
IHC: (+) CD20 (diffuse), CD3 / 5 (stain scattered T cells), EBER-ISH in tumor nuclei, very high Ki67
Tx: rads if herniation imminent, surgery not beneficial, chemo may help
DDx: CNS T cell lymphoma (also perivascular)
Calcifying Pseudotumor of the Neuroaxis (CAPNON)
Can occur anywhere in neuroaxis
- usually appear T1 and T2 hypointense without vasogenic edema, usually have dense calcification, and can have a minimal rim of enhancement on MRI
Usually bland, can have some degeneration and some other junk

Hemangioblastoma

Paraganglioma - Zellballen
SCA3
EBV-assoc DLBCL of CNS (from pathoutllines COW)

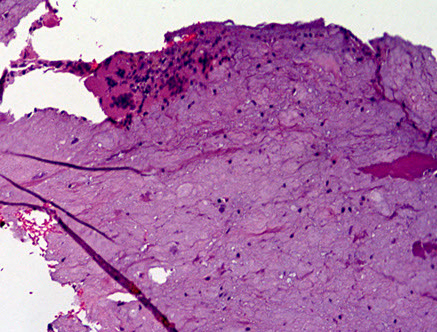
CAPNON
Peripheral Nerve Sheath Tumors
Ganglioneuroma
B9; usually in younger pts (<10 yo) that may exhibit "watery-diarrhea syndrome" from VIP production;
- can also be related to NF1, MEN-IIb, Cowden, TS
- arise in places where sympathetic ganglia are found (usually thoracic, abdominal or adrenal) and are usually well-circumscribed and <15 cm
Micro: made of ganglion cells, some satellite cells, and lots of unmyelinated axons;
- ganglion cells can be pretty dysmorphic (called maturing), and either clustered or scattered with variable Nissl substance
- usually have collagen deposition in stroma, axons and Schwann sheaths bundled in pseudofascicles,
- have uniform maturation (vs ganglioneuroblastomas, which are the less differentiated, but not as undifferentiated as neuroblastoma), and don't have as much neuropil as an immature ganglioneuroma
- intermixed subtype of ganglioneuroblastoma has distinct nests of immature ganglion cells
- usually c some chronic inflam component
IHC: (+) S100, synapto, GFAP (minor)
Genes: usually normal
Tx: Surgery
Ganglioneuroma



Ganglioneuroblastoma
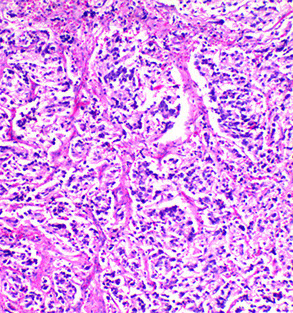
Ganglioneuroblastoma
More undifferentiated form of ganglioneuroma
- looks like it has small round blue cells, almost like inflam
Neuroblastoma - bulky but relatively well-circumscribed solid tumor
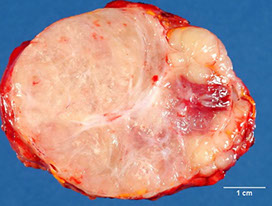
Neoroblastoma of adrenal gland
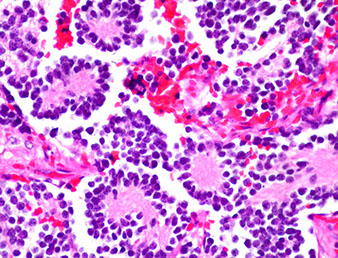
Neuroblastoma
Term applies to a spectrum of neuroblastic tumors including neuroblastoma, ganglioneuroblastomas, and ganglioneuromas
- arise from primitive sympathetic ganglion cells, and have ability to synth and secrete catecholamines (similar to paragangliomas and pheochromocytomas)
- neuroblastomas are ~97% of all neuroblastic tumors, and are fairly unique in the location and biology at presentation and histologic appearance
-- MC in adrenal gland (2/5), abdomen (1/4), thorax (3/20)
- can be assoc c Opsocionus-myoclonus (OMA) and inc VIP secretion as paraneoplastic sundromes
- has hematogenous and lymphatic spread
IHC: (+) SYN, NSE
- neg: vimentin, CD99
Olfactory neuroblastoma (ONB)
- aka esthesioneuroblastoma
ONB is a rare malig tumor of neuroectodermal origin
- present as nasal obstruction, epistaxis, or pain
- clinically is polypoid mass high in nasal cavity
Micro: rich and fragile vascular supply, accounting for its hemorrhagic appearance grossly
- if low grade (well-diff) it is lobulated with transitions to sheets or nests of tumor cells that are round and small c high NC ratio, that have uniform chromatin and small or no nucleoli
- as tumor grade inc, the tumor cells become more pleomorphic c chromatin clumping and prominent nucleoli, and the stroma dec in quantity as tumor is less well-diff
- Homer-Wright pseudorosettes (tumor cells around pink fibrillary material) seen in 1/2 of ONBs, but true (Flexner-type) rosettes c tumor cells around central lumen seen in higher-grade tumors
- high grade tumors also have necrosis, calcs and vascular-lymphatic invasion

Olfactory neuroblastoma (esthesioneuroblastoma)

Neurofibroma
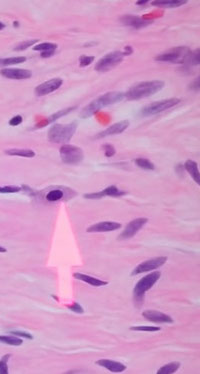
Nerve twig in neurofibroma surrounded by mucin (hyaluronic acid)

Mast cells scattered within a neurofibroma
Plexiform neurofibroma - like a bag of worms
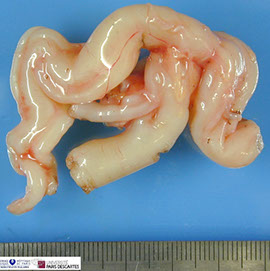
Plexiform neurofibroma

Neurofibroma
Nerve sheath tumor comprising all expected nerve constituents
- solitary tumors not necessarily assoc c NF1; but plexiform and diffuse types more often assoc c NF1 (9/10 and 1/10, respect.)
- 20-40 yo; usually dome-shaped, fleshy lesion protruding from skin from cutaneous nerves, found deep c NF1; usually not encapsulated; has localized, diffuse, and plexiform patterns; mostly sporadic
- usually painless nodules c few sx
Histo: has pale lighter pink color, loose groups of bland, slightly curved spindle cells (Schwann cells) c long wavy nuclei possibly pointed at one end and blunted at one end (like sesame seeds on a hamburger) and scant cytoplasm
- possibly scattered mast cells (look like fried eggs) with delicate granular cytoplasm, which can degranulate and cause inflam
- fibrillary / collagen / mucoid stroma
- can make bluish mucin in background, which is hyaluronic acid
- can have nerve twigs (which are not seen in Schwannoma, which is entirely Scwann cells)
- not encapsulated
- may be very myxoid and confused c myxomas (esp if seen in the extremities)
- can consider malignant change in neutrofibroma if has nuclear atypia (not bad if just by itself), increased cellularity or focal fascicular growth, or inc mits
Localized are most often sporadic in pts that do not have NF1
- much more common than neurofibromas that occur c NF1; usually in pts 20-30 yo
Diffuse MC in head and neck, presents as a plaque-like elevation of skin; is ill-defined and spreads extensively along CT septa and bwt fat cells
"Plexiform" always occurring in childhood, if involves length of nerve - like a "bag of worms" looks tortuous in imaging
- comprises spindled Schwann cells, fibroblasts, capillaries, "shaved-carrot"-like wavy collagen, intermixed axons and mast cells
***are EMA(+) - versus normal neurofibromas
- may result in elephantiasis neuromatosa, where the entire extremity is enlarged
- center of plexiform neurofibroma can turn malignant (to MPNST)
Can be tough to ddx from low-grade MPNST
- look for atypia, inc cellularity, fascicular growth, inc (atypical) mits
- can start to lose or completely lose S100 when transforms
IHC: (+) variable S100 (1/2+), SOX10, focal strong CD34 and Factor XIIIa, scattered neurofilament protein (NFP)
-negative: EMA
DDx: DFSP - can look similar (both are bland, neurofibroma can trap fat like a DFSP), both can stain for CD34, but DFSP will be negative for SOX10 or S100
Px: 2-3% of NF1 pts get malig transformation
Malignant Peripheral Nerve Sheath Tumor
Malignant tumor assoc c peripheral nerve
- most important thing to know is that was derived from a plexiform neurofibroma (or de novo) but not from schwannoma
- assoc c NF1, esp in in kiddos and centrally located (worse px)
- de novo usually in adults in peripheral nerves (better px, 50% 5-yr survival)
- malignant Triton tumor: variant of MPNST with skeletal muscle rhabdomyoblast- differentiation (named after the Triton salamander); this tumor has a very poor px
- MPNST may arise in association with prior radiation therapy. MPNST may be sporadic or arise in association with neurofibromatosis type 1 (NF1) or prior radiation therapy.
Micro: fascicular monomorphic serpentine spindle cells c large vascular spaces with geographic necrosis c palisading around edges
- lots o mits
- may have bone, cartilage or muscle (in 15%), glandular differentiation (on IHC), or melanin
- can have hypercellularity around BVs (Donut sign)
IHC: (+) CD99, S100, CD57, Leu7/CD57, SOX10, p75NTR
-negative: EMA and keratin (usually), H3K27ME3 (tumor cells negative, though endothelial cells and lymphocytes will be positive internal control)
- sensitivity is not very good for SOX10 or S100 (~40%)
A proposed nomenclature for these tumors includes atypical neurofibromatous neoplasm of uncertain biologic potential (ANNUBP), low-grade MPNST, and high-grade MPNST.
- ANNUBP shows at least two of the following four features: cytologic atypia, loss of neurofibroma architecture, hypercellularity, and fewer than 1.5 mitoses/mm2. Lowgrade MPNST has features of ANNUBP but with a higher mitotic index (1.5 - 4.5 mitoses/mm2) and no necrosis. High-grade MPNST shows at least 5 mitoses/mm2 or 1.5 - 4.5 mitoses/mm2 when necrosis is present.
Epithelioid MPNST
Rare (<5%), not assoc c NF1
- not the same as glandular differentiation in conventional MPNST (which can be assoc c NF1 pts)
IHC: (+) S100 (diffuse and strong), EMA / CKs, INI1 loss (2/3)
- neg: melanoma markers
- SMARCB1 gene inactivation is seen in approximately 75% of epithelioid MPNST cases and results in SMARCB1 (INI1) loss by immunohistochemistry.
Genes: complex karyotype, and identification of recurring molecular alterations is not essential to the diagnosis. Frequent and concurrent inactivating mutations have been described in three pathways – NF1, CDKN2A/CDKN2B, and PRC2 core components. Loss of PRC2 core components is a common finding in high-grade MPNST and leads to loss of H3K27me3 expression
Dx if:
- assoc c peripheral nerve
- patchy S-100 +; synovial sarcoma also has S100+ in ~30%
- hx of excised nerve tumor in same area
Has "dumb-bell" shaped morphology in spinal region
Looks "marbled on low-power
DDx: MFH, pleomorphic liposarcoma, synovial sarcoma
MPNST
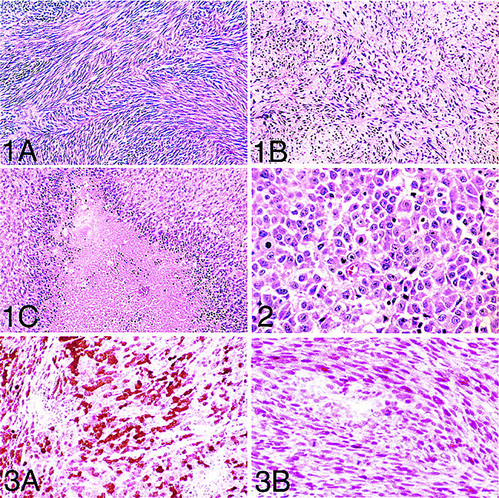
Epithelioid MPNST - strong, diffuse S100
Schwannoma
Nerve sheath tumor consisting solely of Schwann cells that grows as a bulge along a nerve (vs Neurofibroma that looks like the whole nerve enlarged in fusiform shape) - can spare nerve in Schannoma by just cutting off the bulge (not in neurofibroma)
- make up ~1/3 of all spinal tumors; 17/20 cerebellopontine tumors (aka acoustic neuromas) perhaps bc Scarpa's ganglion has the highest density of schwann cells?
- more than 85% of pts have unilateral hearing loss
- pts 20-50 yo; in head and neck, extremity flexors, encapsulated; mostly sporadic
- Cerebello-pontine angle (CPA) tumor DDx: Schwannoma >> Meningioma > Epidermoid cyst
Micro: Well-circumscribed, attached to peripheral nerve (axons should only be seen at the lesion's periphery, if at all)
- Verocay bodies- linear arrays of nuclei and intervening red anuclear stroma
- Antoni A (hypecellualr) and Antoni B (hypocellular) regions
- Hyalinized blood vessels
- Thin fibrous capsule
- when occurs in the GI tract may lack Verocay bodies and Antoni A and B regions
- can auto-infarct, have hemosiderin deposition
IHC: (+) Diffuse, strong S100; pericelllular reticulin positivity, CD34 (in Antoni B region); EMA in periphery of lobules
- negative: NFP
Genes: psammomatous melanotic schwannoma assoc c Carney complex (also assoc c spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and PRKAR1A mutation)
Px: rarely malig transformation
DDx: think of spindle cell melanoma in a spindle cell lesion outside of the retroperitoneum
- Cerebello-pontine angle (CPA) tumor DDx: Schwannoma >> Meningioma > Epidermoid cyst
- Neurofibroma (look like shreeded carrots)
Schwannoma

Psammomatous melanotic schwannoma c a psammoma body, assoc c Carney complex
Traumatic neuroma

Traumatic neuroma
Non-neoplastic, painful mass at site of previous trauma or surgery (esp amputation)
Gross: circumscribed, gray-white nodules, rarely >5 cm
Micro: Disorganized prolif of axonal bundles
- occurs due to distal axonal outgrowth and sprouting in attempt to bridge injured segment and reestablish axonal connection
Is not a Mortons neuroma!
Morton (interdigital) Neuroma
- aka Morton metatarsalgia
- not a true tumor, but a fibrosing process of the plantar digital n that results in praxysmal pain in the sole of the foot
- pain assoc c exercise and alleviated c rest; F>M
- possibly caused by ill-fitting high heels
- surgically see a firm fusiform enlargement of the plantar digital n at bifurcation point
- degenerative changes are the hallmark
Micro: dense perineural and perivascular changes
- elastic fibers decreased at periphery
Neuromuscular Hamartoma
- aka Neuromuscular Choristoma, or Benign Triton Tumor
- rare; tumors made of sk muscle and neural elements collectively cealled "Triton tumors" 2/2 early hypothesis over their pathogenesis
- most well-known is the MPNST c rhadomyoblastic diff (malignant Triton tumor)
- <50 cases, usually in young children
Gross: multinodular; simulate b9 nerve sheath tumors
Micro: highly diff sk muscle fibers which are slightly larger than normal are assoc c small myelinated and nonmyelinated nerves
Perineurioma
Rare nerve sheath tumor in middle aged adults arising in subQ of the limbs coming from perineurial cells (fibroblast-like cells in the perineurium)
Micro: well-circumscribed, no capsule with bland spindle cells with long delicate elongated bipolar cytoplasmic processes in a whorled or storiform architectural pattern;
- common to have predominant myxoid stroma
- should be proliferating lots
Genes: Sclerosing perineurioma assoc c defects in NF2 gene on cr 22 and cr 10 alterations
IHC: EMA+, claudin-1, GLUT1,
- negative S100/NFP - (characteristic!!)
Perineurioma


References
1. Nduom et al. PD-l1 expression and prognostic impact in glioblastoma. Neuro-Oncology 18(2), 195-205, 2016.
2. Osler notes