
Myeloid Neoplasms
Myeloproliferative Neoplasms
Chronic myeloid leukemia (CML), BCR-ABL1-positive
Chronic Neutrophilic Leukemia (CNL)
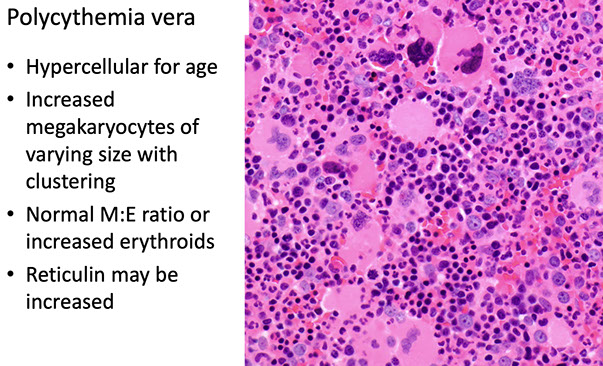
Polycythemia vera (PV)
Primary Myelofibrosis (PMF)
- Prefibrotic / early PMF
- Overt fibrotic myelofibrosis
Essential Thrombocythemia (ET)
Eosinophilia overview *** BONO***
Chronic Eosinophilic Leukemia (CEL), NOS
Myeloproliferative Neoplasms, Unclassifiable
Mastocytosis (not considered an MPN)
- Cutaneous mastocytosis
- Systemic mastocytosis
- Mast cell sarcoma
Myelodysplasic / myeloproliferative neoplasms
Chronic MyeloMonocytic Leukemia (CMML)
Atypical Chronic Myeloid Leukemia (aCML)- BCR-abl1 negative
MDS /MPN with ring sideroblasts and thrombocytosis ( MDS /MPN–RS-T)
Myelodysplastic / Myeloproliferative neoplasm - unclassifiable
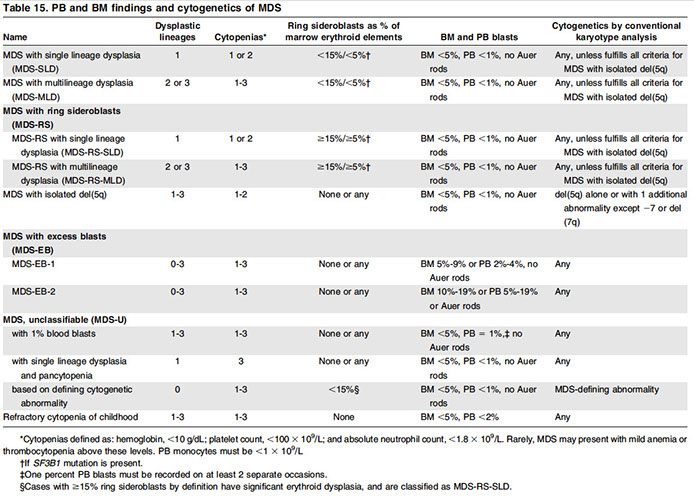
Myelodysplastic Syndromes
MDS with single lineage dysplasia
MDS with ringed sideroblasts (MDS-RS)
MDS with multilineage dysplasia
MDS with excess blasts
- MDS with excess blasts and erythroid predominance
- MDS with excess blasts and fibrosis
MDS with isolated del(5q)
MDS, unclassifiable
Childhood MDS
- Refractory cytopenia of childhood
Acute myeloid leukemia (AML) and related precursor neoplasms
Acute myeloid leukemia (AML) with recurrent genetic abnormalities
- Acute myeloid leukemia with t(8;21)(q22;q22.1); RUNX1-RUNX1T1
- Acute myeloid leukemia with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11
- Acute promyelocytic leukemia (APML) with PML-RARA
- Acute myeloid leukemia with t(9;11)(p21.3;q23.3); KMT2A-MLLT3
- Acute myeloid leukemia with t(6;9)(p23;q34.1); DEK-NUP214
- Acute myeloid leukemia with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM
- Acute myeloid leukemia (megakaryoblastic) with t(1;22)(p13.3;q13.1); RBM15-MKL1
- Acute myeloid leukemia with BCR-ABL1
-
Acute myeloid leukemia with gene mutations
- Acute myeloid leukemia with mutated NPM1
- Acute myeloid leukemia with biallelic mutation of CEBPA
- Acute myeloid leukemia with mutated RUNX1
Acute myeloid leukemia with myelodysplasia-related changes
Therapy-related myeloid neoplasms
Acute myeloid leukemia, not otherwise specified
- Acute myeloid leukemia with minimal differentiation
- Acute myeloid leukemia without maturation
- Acute myeloid leukemia with maturation
- Acute myelomonocytic leukemia
- Acute monoblastic and monocytic leukemia
- Pure erythroid leukemia
- Acute megakaryoblastic leukemia
- Acute basophilic leukemia
- Acute panmyelosis with myelofibrosis (APMF)
Myeloid sarcoma
Myeloid proliferations associated with Down syndrome
- Transient abnormal myelopoiesis associated with Down syndrome
- Myeloid leukaemia associated with Down syndrome
Leukemia cutis - see Neoplastic Dermatology
Myeloid / lymphoid neoplasms with eosinophilia and gene rearrangement
Myeloid / lymphoid neoplasms with PDGFRA rearrangement
Myeloid / lymphoid neoplasms with PDGFRB rearrangement
Myeloid / lymphoid neoplasms with FDFR1 rearrangement
Myeloid / lymphoid neoplasms with PCM1-JAK2
Blastic plasmacytoid dendritic cell neoplasm
Myeloid neoplasms with germline predisposition
Acute leukemias of ambiguous lineage
Myeloid neoplasms with germline predisposition without a pre-existing disorder or organ dysfunction
- Acute myeloid leukaemia with germline CEBPA mutation
- Myeloid neoplasms with germline DDX41 mutation
Myeloid neoplasms with germline predisposition and pre-existing platelet disorders
- Myeloid neoplasms with germline RUNX1 mutation
- Myeloid neoplasms with germline ANKRD26 mutation
- Myeloid neoplasms with germline ETV6 mutation
Myeloid neoplasms with germline predisposition associated with other organ dysfunction
Myeloid neoplasms with germline GATA2 mutation
Myeloid neoplasms with germline predisposition associated with inherited bone failure syndromes and telomere biology disorders
Acute undifferentiated leukaemia
Mixed-phenotype acute leukaemia with t(9;22)(q34.1 ;q11.2); BCR-ABL1
Mixed-phenotype acute leukaemia with t(v; 11 q23.3); KMT2A-rearranged
Mixed-phenotype acute leukaemia, B/myeloid, NOS
Mixed-phenotype acute leukaemia, T/myeloid, NOS
Mixed-phenotype acute leukaemia, NOS, rare types
Acute leukaemias of ambiguous lineage, NOS
Overview of Myeloid Neoplasms
Nonblasts sometimes considered "blast equivalents"
- promonos in dx of acute monocytic or myelomonocytic leukemia
- Promyelos in dx of acute promyelocytic leukemia (APML)
- erythroblasts in "pure erythroleukemia"
All cases should get cytogenetic culture
- morphology guides use of specific molecular assays
Sx: most have cytopenias, dyspoiesis, and can develop acute leukemia
- present c fatigue, infx, or bleeding
- S-megaly usually not present

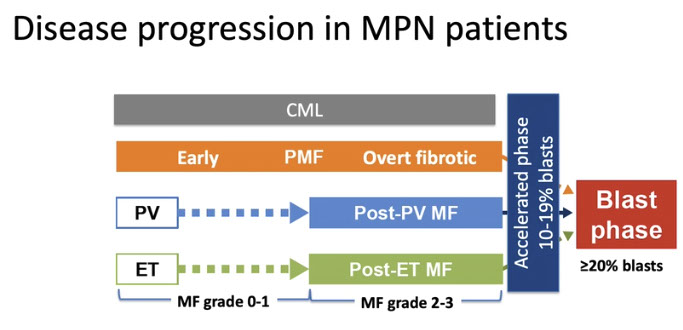
Myeloproliferative Neoplasms
Are clonal hematopoietic stem cell disorders characterized by proliferation in the bone marrow of one or more of the myeloid lineages
Usually >1 myeloid lineage is involved
- Mild to marked erythrocytosis, leukocytosis, and thrombocytosis
- Most patients may be diagnosed with a PB smear.
Varying degrees of marrow fibrosis
- Splenomegaly
Peak incidence is in the fifth to sixth decades of life, although may occur in children
MPN overview [2]


Chronic myeloid leukemia (CML), BCR-ABL1 positive
_____________________
Leukocytosis, with immature and mature cells. Thrombocytosis is common, S-megaly from EMH
- can be dx'd from PB, but should get BM to ensure enough material for KT and confirm phase of dz
t(9;22) creates the Philadelphia chromosome (Ph1) and the BCR/ABL1 chimeric gene, the molecular basis of the disease
- Detectable by cytogenetics (karyotype, FISH) or molecular genetics (RT-PCR)
Other cytogenetic abnormalities (+Phl, +8, iso17q) show disease progression
Disease of adults (but reported in all ages).
_____________________________________
_____________________________________
Sx: Fatigue, shortness of breath on exertion, malaise, fullness of upper abdomen, anorexia and weight loss, priapism: due to extremely elevated WBC or platelet count.
_____________________________________
Peripheral Blood Lab Findings:
Marked leukocytosis (50 - 600 X 10^9/L). Basophilia. and eosinophilia
Thrombocytosis.
Anemia is normochromic normocytic.
Elevated serum vit B12 and vit B12 carrying capacity
- Decreased leukocyte alkaline phosphatase (LAP)
- derived from precursor cell to myeloid and lymphoid lineages
Marked myeloid hyperplasia with “myelocyte bulge”.
BM Findings
- markedly hypercellular (~100% cellularity)
- markedly elevated M:E ratio
Megakaryocytic hyperplasia and clustering.
-- "Dwarf mgkcs" - lots o small hypolobated megs
Myelodysplasia is not a major feature.
Marrow fibrosis (reticulin fibrosis) is variable
"pseudo-Gaucher" cells - see below
Bi-/triphasic: Chronic Phase (CP), then Accelerated Phase (AP) or Blast Phase (BP)
- stays in chronic phase for ~3.5 yrs; half have an accelerated phase that can progress to blast phase
_____________________________________
Chronic Phase (CP)
Leukocytosis w myelocytes and segs. Eosinophilia common
- blasts ususally <2% on PS
- marrow hypercellular w myeloid lineage inc and dwarf megakaryocytes (small + hypolobated)
-- BM fibrosis in 30% (get reticulin stain??), which is assoc w inc megakaryo # and poor prog
-- may see Gaucher cells and sea-blue histiocytes in BM too
Tx: Protein Kinase Inhibitor (PTKI) inhibitor therapy causes dec myeloid cells, better mega poiesis, dec fibrosis, dec prolif
_____________________________________
Accelerated Phase (AP)
Becoming less common in the age of TKI therapy, no universally accepted criteria to dx AP of CML
- see table 2 above for a complete list of dx criteria
- criteria include hematologic, morphologic and cytogenetic parameters in addition to genetic evolution and evidence of resistance to TKIs
1) persistent/ increasing WBC or splenomegaly
2) persistent thrombocytosis uncontrolled by therapy
3) persistent thrombocytopenia unrelated to therapy
4) colonal cytogenetic evolution occuring after initial diagnostic karyotype
5) >20% basophils in PB
6) 10-19% myeloblasts in PB or BM
- 1-4 --> CP to AP; 5/6 --> AP to BP
_____________________________________
Blast Phase (BP)
1) blasts > 20% PB WHB or nucleated cells on BM
- large focal blast collection may clinch BP dx even if total blast count < 20%
2) extramedullary blast proliferation
-- CD34 or TdT helps to identify blasts, but not always...
70% are myeloid (AML), 30% lymphoid (B-ALL)
- the lymphoid BP cases are usually precursor B cell lineage, but usually express myeloid markers as well
_____________________________________
Genetics
95% w/ t[9:22], bcr-abl, on the Philadelphia chromosome
- BCR (breakpoint cluster region) gene on 22q11.2 fuses c ABL (Abelson, for a leukemia virus that carries similar protein) gene on 9q34
-- may have cryptic fusion requiring FISH
- this usually causes inc tyrosine kinase activity
- "metamyelocyte bump": cells normally decrease in number from most to least mature; but get a strange bump in the metamyelocyte cell line here
p190 - ALL
p210 - CML, sometimes ALL
p230 - CML c chronic neutrophilia and tbcytosis
Cytogenetics: t(9;22) on conventional karyotype
FISH: Fusion of BCR and ABL1 probes
Moelcular: RT-PCR demonstration of BCR-ABL1 fusion
Rare cases may have cytogenetically cryptic BCR-ABL1 rearrangement
_____________________________________
DDx: - Myelofibrosis with myeloid metaplasia.
- Infection or reaction to tissue necrosis or neoplasm (leukemoid reaction)
- Leukocyte (or neutrophil) alkaline phosphatase (LAP/NAP):
- Normal or increased in leukemoid reaction.
- Decreased in CML.
_____________________________________
IHC: Low leukocyte alkaline phosphatase (vs leukemoid reaction) in CP
_____________________________________
Px
Inexorably progress to an acute leukemia with loss of differentiation ('blast crisis' or 'blast phase')
- AML types:
70% myeloid,
30% B-lymphoid (underscores pluripotent nature of the CML stem cell)
If transforms to ALL AML ("blast crisis") = Acute Progression = Rapid Death (20%/year)
If untreated:
Proliferative and symptomless phase - 6.3 years.
Preclinical - 19 months.
Terminal, advanced - 3 years mean survival.
Chronic phase (5-10%) -> Accelerated phase (11-19%) -> Blast crisis (>20% blasts)
~2/3 myeloid (AML)
~1/3 lymphoid (ALL)
_____________________________________
Tx: Hydroxyurea, alpha-interferon
- Bone marrow transplantation (BMT)
- Gleevec (imatinib) and Sprycel (desatinib) - inhibit tyrosine kinase activity of chimeric BCR/ABL protein
***Imatinib ( *** "I'm a in Philadelphia!" says Becker. ***) IMitates ATP and competes for binding site on BCR-ABL1 kinase domain and prevents phosphorylation of tyrosine residues on its substrates
- BMT is a cure (but not the best initial tx)
____________________________________
Px: may live normal lifespan, but should be regularly monitored for BCR-ABL1 burden and for evidence of resistance to TKI therapy
- inexorably progress to an acute leukemia with loss of differentiation
- Acute leukemia types: 70% myeloid, 30% B-lymphoid (the occurence of B-lymphoid blastic progression underscore the pluripotent nature of the CML stem cell)


CML- chronic phase PB

CML - Marrow fibrosis

CML: FISH major breakpoint; Red=ABL, Green = BCR





CML - dwarf megs


TKIs in CML
Chronic Neutrophilic Leukemia (CNL)
_____________________________________
Rare, in older males, always in PB and BM, usually infilatrates liver and spleen
- Reduced numbers of CFU-C (colony-forming units).
No cytogenetic abnormality found.
Persistent leukocytosis without a left shift (100 X 109/L).
- Toxic granulation and Döhle bodies are present.
- Rare NRBC can be seen.
- Elevated LAP.
_____________________________________
BM marked cellularity.
Neutrophilic hyperplasia.
No evidence of myelodysplasia.
Negative for Ph1 chromosome.
Neutrophils often appear toxic with coarse granules
_____________________________________
Diagnostic criteria:
1.) WBC > 25,000 (in PB), with majority (>80%) neutrophils + bands, and <10% immature myelocytes (pro-,myelo-,metamyelo-), <1% blasts
2.) Hypercellularity on BMBX, c inc neutros (which mature normally), normal blasts and Mkcs
3.) Hepatosplenomegaly
4.) No underlying cause for neutrophilia (infx or tumor)
5.) No Philadelphia chromosome or BCR-ABL1 fusion genes
6.) No PDGFRA, PDGFRB, or FGFR1 rearrangements
7.) No polycythemia vera, primary myelofibrosis, or essential thrombocythemia
8.) No MDS, MPN, granulocytic dysplasia
9) CSF3R T618I or other activating CSF3R mutation
Must exclude other myeloproliferative neoplasms, myelodysplasia, and anything that can normally cause neutrophilia
- since neutrophilia often occurs with multiple myeloma, BM should be examined for plasma cell prolif (in olden days, chronic neutrophilia assoc c plasma cell neoplasms, but the clonality of neutrophils was never tested, so most likely was 2/2 reactive neutrophilia from cytokine production and not a clonal neoplasm)
Should (in ~90%) have the CSF3R mutation described above
- pts will occasionally be JAK2 positive
One pt has been successfully tx'd c imatinib who had t(15;19)(q13;p13.3), meaning some cases may have fusion genes
_____________________________________
Px: ranges 6 months to 20 years
- is a slowly progressive disorder, which may eventually cause anemia and thrombocytopenia
- some patients may get MDS which progresses to AML (may have been caused by cytotoxic tx)
Revised criteria for dx of CNL




Mutation of the TROR receptor itself is due to mutations of the MPL gene, which encodes it [2], CALR is response for the folding of the MPL protein

MDS 5q del, CMML, MDS/MPN-RS-T, AML inv 3
PV in PB

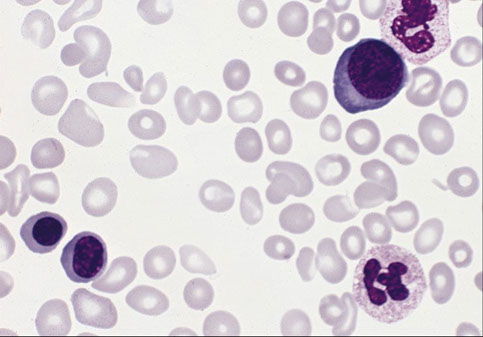
PV with marrow fibrosis

Polycythemia Vera (PV)
_____________________________________
Panmyelosis and specifically increased red cell mass (RCM)
RBC count: - >5.9 X 1012/L in women; >6.6 in men
- Extremely high erythrocyte count (may reach 10 X 1012/L).
Hgb = males >17.5 g/dL, females >15.5 g/dL.
Hct = males >55% , females >47%.
- Mean Cell Volume (MCV) and Mean Cell Hemoglobin Concentration (MCHC): low normal to low because of absent or decreased iron stores
- chronic bleeding (abnormal platelet function)
-therapeutic phlebotomy
- increased RBC production and turnover
Mild granulocytosis (may go as high as 50 X 109/L) and thrombocytosis
Basophils are elevated
Molecular basis is JAK2 (Janus kinase 2) gene mutations (usually JAK2 V617F), >98% patients
____________________________________
PB findings:
- Normoblasts may be seen
- Reticulocytes are not significantly increased
- Difficult to prepare a good PB film due to high viscosity (For better smear - mix equal parts of blood specimen and normal saline)
- Increased serum vitamin B12 and serum vitamin B12 binding capacity
- Increased leukocyte alkaline phosphatase (LAP)
BM
Hypercellular with increased normoblasts and megakaryocytes
Fibrosis worsens as disease progresses
Iron is decreased or absent
____________________________________
Labs
Decreased serum iron and ferritin.
Decreased erythropoietin levels.
____________________________________
Sx: Headaches, vertigo, ringing in the ears, blurred vision, itching eyes, upper GI pain (may be secondary to peptic ulcers), pruritis (red, itchy skin).
- Thromboses, either venous or arterial, are common (strokes)
- Most patients are 50 to 60 years, but all age groups
- Splenomegaly and hepatomegaly due to EMH (extramedullary hematopoiesis)
- Gout and hypertension.
_____________________________________
3 phases:
1.) prodromal - only has mild erythrocytosis
2.) polycythemic stage - definite increase in RBCs
3.) terminal stage - lots of BM fibrosis, hypersplenism and EMH
May eventually progress to MDS or AML; Hgb normal to dec in later stages
- reticulin stain helps visualize marrow fibrosis in late stage
Must exclude other causes of polycythemia
Survival >10 years common
- most pts die from thrombosis or hemorrhage, ~20% get MDS or AML (which increases with use of certain cytotoxic therapies)
_____________________________________
Diagnostic criteria
(need all 3 major or 2 major and the minor)
Major Criteria:
1.) Hgb > 16.5 Male; >16 Female, or
Hct > 49 M, >48 F, or
Increased RCM
2. Hypercellular Bmbx with trilineage growth (panmyelosis) with pleomorphic mature megs (different sizes)
3. Presence of JAK2V617F or JAK2 exon 12 mutation
Minor criteria:
Low EPO level
_____________________________________
DDx: PV van mimic ET in early stages
_____________________________________
Genes
JAK2 in >95% of PV patients, but is non-specific
- nearly all pts that progress to MDS of AML have a cytogenetic abnormality (ie +8, +9, del(20q), del(13q), del(9p))
- Jak-2 protein stimulates STAT pathway
- MC JAK2 mutation is G to T substitution on nucleotide 1849, causing val to phe substitution at codon 617 (Val617Phe)
- less commonly can have 2nd activating mutation c JAK2 at exon 12
- no MPL mutations assoc c PV
Long term - tear drop cells (dacryocytes) secondary to myelofibrosis
- PV transforms into AML in a significant number (may be secondary to therapy)
- Without treatment median survival is ~18 months.
- With treatment survival varies from 8.9 to 13.9 years, depending on the therapy.
_____________________________________
Post-PV MF
Diagnostic criteria
Required criteria
1.) Previously diagnosed PV
2.) Significant marrow fibrosis
Additional criteria (need at least 2)
1.) Anemia
2.) PB leukoerythroblastic
3.) Splenomegaly
4.) at least 1 of 3 constitutional sx: weight loss, night sweats, fever
_____________________________________
Tx: Phlebotomy to reduce blood viscosity
- 32P or alkylating chemotherapy to reduce bone marrow production - increased risk of secondary acute leukemias and lymphomas
- Hydroxyurea, alpha-interferon
- Aspirin to prevent clots
_____________________________________
Px
Thrombotic complications are more common than in ET
- excellent long-term survival
- may develop fibrosis over time (in burnt out phase)

[2]




PMF
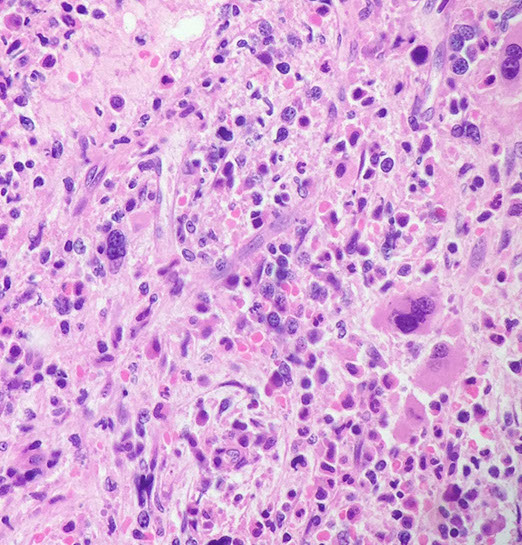
Primary Myelofibrosis
Primary Myelofibrosis (PMF)
_____________________________________
aka
(agnogenic myeloid metaplasia, AMM) (idiopathic myelofibrosis, IMF) (myelofibrosis with myeloid metaplasia, MMM) (chronic idiopathic myelofibrosis, CIM)
_____________________________________
Challenges of diagnosis PMF[2]:
- in fibrotic phases of disease, must distinguish from other causes of bone marrow fibrosis
-- close attention to morphology and mutation status (JAK2/MPL/CALR) is helpful
- in early phases of disease, fibrosis is minimal or absent
- correct diagnosis is important, since early PMF has a high risk of progression to a fibrotic disease phase and has worse prognosis than other MPNs
- morphologically distinct from ET; erythrocytosis is absent
-- increased bone marrow cellularity and more frequent, clustered megakaryocytes in early PMF compared to ET
_____________________________________
- Characterized by fibroblast proliferation, increased collagen, fibrosis, and granulocytic hyperplasia in BM; extramedullary hematopoiesis (metaplasia) in spleen and liver
- Fibrosis is a secondary reaction (mgkcs and platelets contribute to fibrosis).
- Middle aged to older people, rarely in children.
- Defective platelets (dysplastic megakaryocytopoiesis) and early cell death.
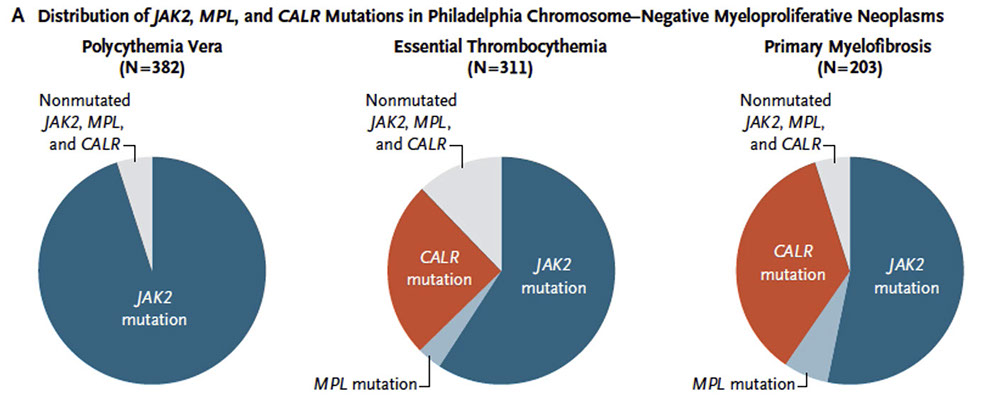
Dz assoc c JAK2 gene mutations (~50% pts) and MPL and CALR gene mutations
Anemia, abdominal pain, indigestion, fullness after eating (splenomegaly), anorexia and weight loss, fever, night sweats, lethargy and weakness.
- may look ashen; have HSmgaly, petechiae, epistaxis
Dz has long peaceful periods marked by life-threatening hemorrhagic or thrombotic events
_____________________________________
PB findings
- Mild normochromic, normocytic anemia
- Dacryocytes (cells’ tortuous circulation through enlarged spleen)
- Occasional NRBC, mild reticulocytosis.
- Giant, agranular platelets
- Rare megakaryocyte fragments.
- Occasional immature myeloid cells
- Rare myeloblasts
- Elevated serum vitamin B12 and vitamin B12 carrying capacity
- Variable (normal to increased) LAP
BM findings
“Dry tap” - no aspirate.
- Reticulin fibrosis and collagen deposition
- Hypocellularity
- Increased number of megakaryocytes
_____________________________________
Can mimic ET and PV, BMBx helps to differentiate
- morphologic features (dacryocytes in PB) can help differentiate, but are variable
_____________________________________
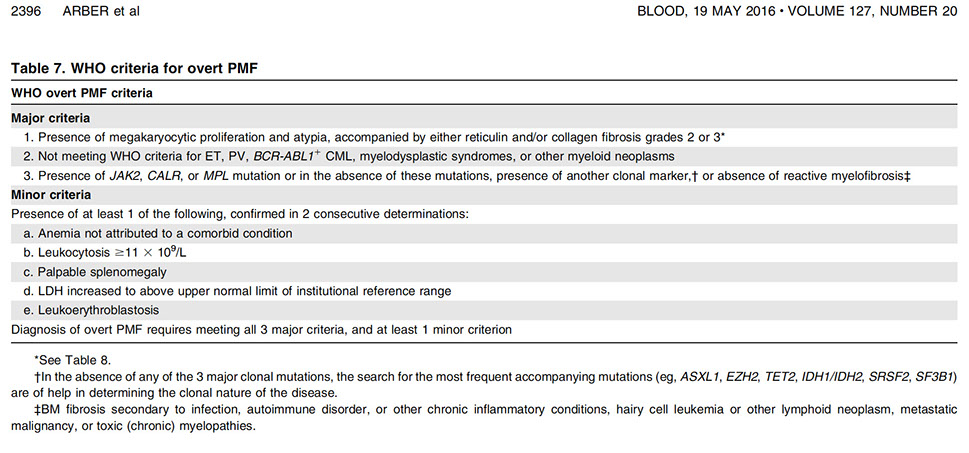
Dx Criteria (need all 3 major and 2 minor):
Major
1.) Mkc prolif + atypia, c fibrosis (if no fibrosis, then need hypercellular BM)
2.) No PV, BCR-ABL1 CML, MDS or other neoplasm
3.) (+) JAK2 V617F or other marker (ie MPL W515K/L)
Minor
1.) Leukoerythroblastosis 2.) Inc serum LDH 3.) Anemia 4.)S-megaly
_____________________________________
Genetics:
del(13)(q12-22) or der(6)t(1;6)(q21-23;p21.3) is very suggestive but not diagnostic of PMF
- del(20q) and partial trisomy 1q are the MC recurrent abnormalities
_____________________________________
Px
Survival from months to decades depending on the stage at first diagnosis
- worse Px in pts >70 yo, Hgb < 10, plt < 100k, and abnormal karyotype
Death from BM failure, portal HTN, thrombosis, CHF and AML
- AML may be part of the natural progression of the dz
Hemorrhage, infection, or cardiac complications are the immediate cause of death (cytoreduction and phlebotomy may help this)
- can develop fibrosis over time ("Burnt-out phase")
- 5%-8% transform into AML.



PMF advanced stage with blue collagen fibrosis, osteosclerosis


Intrasinusoidal hematopoiesis with sinus megakaryocytosis is a hallmark of this disease [2]
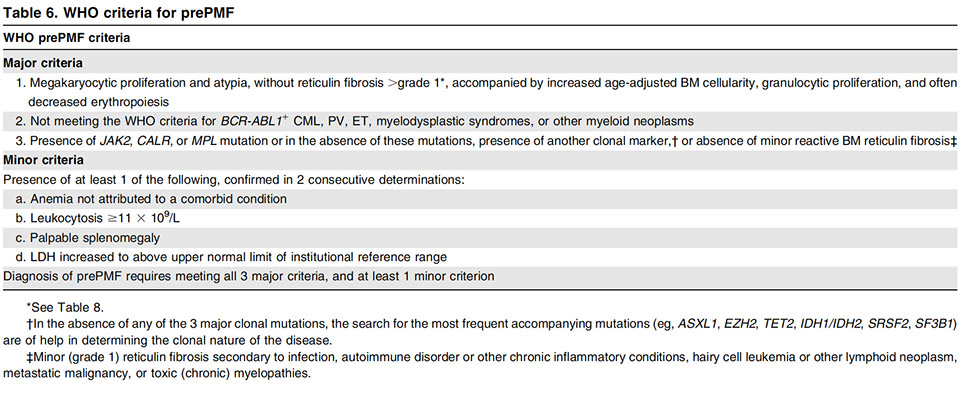
ET vs PMF [9]

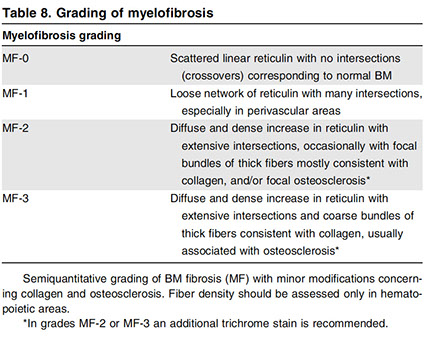

FIGURE 6 Algorithm for distinguishing essential thrombocythemia (ET) from pre–primary myelofibrosis (PMF). LDH, lactate dehydrogenase; MPN,U, myeloproliferative neoplasm, unclassifiable. In cases with thrombocytosis and JAK2 mutation, first exclude polycythemia vera by using World Health Organization 2016 criteria. This conservative approach prevents false-positive diagnosis of PMF, but rare early cases of pre-PMF may be missed and called ET or MPN,U. aIf a diagnosis of MPN,U is rendered, we recommend repeat biopsy in one year. [9]
Distiguishing PMF, AIMF, and MDS-F [9]

Essential Thrombocythemia (ET)
_____________________________________
- Thrombocytosis in excess of 1000 X 109/L
- Spontaneous aggregation of functionally abnormal platelets
- No apparent cause for thrombocytosis
- Increase in platelet mass
- Disease associated with JAK2 gene mutations (~50% patients have them) and MPL and CALR gene mutations
- Second least common MPN.
_____________________________________
Dx criteria (must have all 4):
1.) Plt > 450k for duration of dx process
2.) Mkc prolif on bmbx; no inc/left-shifted granulo-/erythropoiesis, only very rarely has inc reticulin fibrosis
3.) No PV, PMF, BCR-ABL1 CML, MDS or other neoplasm
4.)(+) JAK2 V617F, or if negative, no other reason for reactive thrombocytosis
Usually in older pts, but second peak in 30 yo F; though some pts may show vascular occlusion (ie Budd-Chiari) or hemorrhage (mucosal)
Epistaxis, vomiting of blood, easy bleeding.
- Thrombotic events are common.
- Fatigue; HSmegaly; Priapism (arterial or venous thrombosis); Pulmonary emboli; Gangrenous toes
No EMH in spleen, although may sequester platelets
_____________________________________
PB findings
Thrombocytosis 1000 X 10^9/L.; large platelet aggregates
Platelet anisocytosis; bizarre forms (agranular, pseudopods)
- Occasional megakaryocyte fragments
- Target cells, acanthocytes, and Howell-Jolly bodies if splenic infarction.
Coagulation studies: abnormal platelet aggregation
- will not see leukoerythroblastosis or dacryocytes
BM findings
- May have poor aspirate - myelofibrosis.
- Hypercellular marrow with mgkc hyperplasia.
- giant Mkc's c staghorn nuclei, hyperlobulated, lots of cytoplasm, but not as bizarre as in PMF
- no inc blasts, dysplasia, or significant fibrosis
~ Half have JAK2 V617F mutation
Dz has long peaceful periods marked by life-threatening hemorrhagic or thrombotic events
- Few patients progress to MDS or AML, and is usually a results of therapy
Life expectancy is near normal bc dz dx'd later in life
_____________________________________
DDx
Secondary thrombocytosis:
- Normal platelet aggregation studies.
- No hepatosplenomegaly
<1000 X 109/L plts.
Causes: infection, inflammation, neoplasms, splenectomy.
__________________________________________
Px: occasional thrombotic or bleeding outsomes
- excellent long-term survival,
- do not develop marrow fibrosis
- <5% progression to AML (little if any increase than the general population)



ET - peripheral smear


Essential Thrombocythemia
Megs look like staghorn nuclei in the biopsy, and like a bunch of grapes in the aspirate [2]


Eosinophilia
_____________________________________
• Group of disorders marked by sustained overproduction of eosinophils
• Eosinophilic infiltration and release of mediators may cause damage to multiple organs
• Variable cytologic features
Numerous causes, both neoplastic and non-neoplastic
• Broad workup can be daunting
• Methodical algorithmic approach
– Manage ancillary testing
– Ensure thorough, appropriate workup
– Diagnose/exclude specific disease entities
Consensus panel 2012, Valent et al.
Hypereosinophilia (HE)
• Persistent eosinophilia >1.5 x 109/L on 2 separate examinations (> 1 month) and/or
• Tissue HE defined by >20% eosinophils in bone marrow, and/or extensive tissue infiltration determined by a pathologist, and/or marked deposition of eosinophil granules and proteins in tissue
Hypereosinophilic syndrome (HES)
• Criteria for HE fulfilled and organ damage/dysfunction due to HE and must exclude other reasons for organ damage
- DDx: CEL,NOS has increased blasts (>2% in PB, >5% in BM; <20% in both) or a clonal cytogenetic abnormality
Complication:
-- thrombotic events can occur, requiring amputation
_____________________________________
Subtypes of Hypereosinophilia
• Hypereosinophilia (HE)
– HEFA (familial), HEUS (undetermined significance), HEN (neoplastic), HER (reactive)
• Hypereosinophilic syndrome (HES)
– HEI (idiopathic), HEN, HER
• Eosinophil-associated single-organ diseases
– Specific syndromes accompanied by HE: e.g. EGPA
– Other conditions accompanied by HE: e.g. eosinophilic esophagitis
FA, familial; US, undetermined significance; N, neoplastic; R, reactive; I, idiopathic
_____________________________________
Approach to Eosinophilia
• Systematically collect data!
• Exclude secondary causes
• Assess for a primary clonal disorder
– Specific treatments for certain diseases (eg, TKI in CML, PDGFRA-rearranged)
• Idiopathic HE/HES is a diagnosis of exclusion
- Can be fairly overwhelming as to how and where to begin
• Cases are uncommon too
• Clinical practice guidelines/algorithms
– Reiter A, Gotlib J: Myeloid neoplasms with eosinophilia. Blood. 2017; 129: 704–714
– Wang SA. The Diagnostic Work-Up of Hypereosinophilia. Pathobiology. 2019;86(1):39–52
– Boyer DF. Blood and Bone Marrow Evaluation for Eosinophilia. Arch Pathol Lab Med. 2016;140(10):1060–1067
– Butt NM, et al. Guideline for the investigation and management of eosinophilia. Br J Haematol. 2017;176(4):553–572
– Fang H, et al. A Test Utilization Approach to the Diagnostic Workup of Isolated Eosinophilia in Otherwise Morphologically Unremarkable Bone Marrow: A Single Institutional Experience. Am J Clin Pathol. 2018;150(5):421–431
Approach to PB and BM Eosinophilia
• Helpful if clinical history, lab studies provided
– Skin lesions, tryptase, splenomegaly, known diagnosis…
• Eosinophil cytology
– Normal: 2-3 lobes, red-orange large granules pack the cytoplasm
– Abnormal: Alterations in segmentation and granulation, vacuolization
– Abnormal cytologic features are seen more often in primary clonal states but are not exclusive
– Eosinophil cytology (abnormal) should not be used alone to indicate clonal eosinophilic disease
Assess for additional pathologic clues
– PB: Monocytosis, dysplasia, increased or abnormal blasts, basophilia
– BM: Cellularity, megakaryocyte morphology, fibrosis, dysplasia, abnormal mast cells/clusters, lymphoid infiltrates, granulomas, metastasis, organisms
_____________________________________
Hyper-IgE syndrome
Triad of eczema, recurrent skin infections, and elevated IgE levels
- pathogenesis due to skewing of TH2 differentiation, which produce increase IL-4, IL-5, and IL-13
- most common form of autosomal dominant hyper-IgE syndrome results from dominant negative mutations STAT3
- shares some features wiht Wiscott-Aldrich syndrome
LASOP lecture, Dr. Reichard, 1/18/2020



Distinguishing HES from CEL (WHO 2016)

Drug Rash with Eosinophilia and Systemic Symptoms (DRESS)
Originally described in response to phenytoin, usually occurs 2-6 weeks after drug exposure
- patients have a rash, in addition to lymphadenopathy and fevers
reactivation of herpesvirus (esp HHV6) has been implicated in the pathogenesis
May mimic a hematologic malignancy!!
- may show increased Langerhans cells, similar to LCH
- can be in lymph nodes with very high CD30 cells! (see image)
Micro:
- high fatality rate (10%) usually due to fulminant hepatitis or macrophage activation syndrome




DRESS mimicking LCH. Case 229 also showed expansion/effacement of the paracortex (D, ×10) by cells with grooved nuclei consistent with Langerhans cells (E, ×40) confirmed by CD1a immunohistochemistry (F, ×10). [4]
Reactive lymphadenopathies in drug reaction with eosinophilia and systemic symptoms. A-C, Case 262 showed paracortical expansion of lymphocytes and histiocytes (A, ×20), with CD3 showing a predominance of T cells (B, ×20) and CD30 highlighting increased immunoblasts (C, ×20).
1 - 2
<
>
Lymphocytic variant of hypereosinophilic syndrome (LV-HES)
• Cytokine-driven eosinophilia due to overproduction of eosinophil growth factors by T-cells
• CD3(-), CD4(+), CD5(+), CD7(-) phenotype, but criteria for peripheral T-cell lymphoma is not met
• Often a minority of circulating cells (<20%)
• Cutaneous manifestations
• Often responsive to steroid therapy (try to eliminate the T-cell clone)
Updates in Eosinophilia
• Myeloid/Lymphoid neoplasm with eosinophilia with recurrent genetic abnormality
• Distinct category originated in the WHO classification in 4th edition (2008)
• Recently expanded to include PCM1-JAK2 in the revised 4th edition (2017)
• Increasing recognition of additional genetic variants
Beware of diagnostic pitfalls in eosinophilic disorders
• Morphologically occult/subtle infiltrates
• Abnormal eosinophil cytology is not synonymous with clonal disorder of eosinophils
• Absence of eosinophilia to alert one to a recurrent genetic abnormality
• Atypical presentation (extramedullary, lymphoblastic leukemi/lymphoma)
• Cytogenetically cryptic cases
Lymphocytic variant of hypereosinophilic syndrome (LV-HES)
.jpg?crc=190692539)
Idiopathic Hypereosinophilic Syndrome (IHES)
_____________________________________
• Diagnosis of exclusion
– All reactive/secondary causes of hypereosinophilia
– Lymphocyte-variant of HES
– CEL, NOS
– WHO-defined myeloid malignancies (eg, AML, MPN)
– WHO-defined MLNE with eosinophilia with PDGFRA, PDGFRB, FGFR1 and PCM1-JAK2
__________________________________________
Px:
Generally does better than CEL

Hypercellular bone marrow with panhyperplasia; All ancillary studies are negative... Diagnosis? = Idiopathic hypereosinophilic syndrome (IHES)

Chronic Eosinophilic Leukemia (CEL) - not otherwise specified
_____________________________________
- Autonomous, clonal proliferation of eosinophilic precursors in PB, BM, and peripheral tissues
- Analogous to CML or CNL
- Eosinophilia (>1.5x109/L) in PB
and...
(- Blasts increased in PB (2%-19%) and/or in BM (5%-19%)
or
Clonal abnormality
- Cytogenetic: +8, iso(17q)
- Molecular genetic: JAK2 occasionally
- Excluding BCR/ABL1, and rearrangements of PDGFRA, PDGFRB, and FGFR1 )
_____________________________________
Diagnostic criteria:
1. Eosinophilia (eosinophil count > 1.5 x 109/L)
2. WHO criteria for BCR-ABL1 positive chronic myeloid leukemia, polycythemia vera, essential thrombocythemia, primary myelofibrosis, chronic neutrophilic leukemia, chronic myelomonocytic leukemia and BCR-ABL1 negative atypical chronic myeloid leukemia are not met
3. No rearrangement of PDGFRA, PDGFRB or FGFR1, and no PCM1-JAK2, ETV6-JAK2, or BCR-JAK2 fusion
4. Blast cells constitute <20% of the cells in the peripheral blood and bone marrow, and inv(16)(p13.1q22), t(16;16)(p13.1;q22), t(8;21)(q22;q22.1), and other diagnostic features of acute myeloid leukemia are absent
5. There is a clonal cytogenetic or molecular genetic abnormality“ OR blasts account for >2% of cells in the peripheral blood or > 5% in the bone marrow
BM hypercellular c eos, normal blast #'s (up to 20%); Charcot-Leyden crystals are present; fibrosis is common (from eo degranulation)
- dysplastic megakaryocytes also common in CEL ;7]
Leukemic infiltration and release of eo contents can cause organ damage
Blast counts should be <20% w/o BCR-ABL1, Ph chromosome or PDGFRA
HyperEosinophilic Syndrome (HES) is persistent eosinophilia for 6 months w/o organ damage or evidence of eosinophilic clonality
Occurs mainly in adult men who are asymptomatic, but may have pruritis, angioedema, fatigue, diarrhea
DDx
must exclude Reactive eosinophilias (***NAACP: Neoplasia, Allergies, Asthma, Collagen vascular diseases, Parasites), Medications,
- Hypereosinophilic Syndrome (HES; Persistent eosinophilia; Blasts not increased in PB or BM; No evidence of clonality);
- Loeffler's syndrome, angiolymphoid hyperplasia, Kiruma's disease
- other neoplasms that release IL's (Hodgkin's, SM) must be excluded
Border of IHES and CEL, NOS: Role of Morphology
• Study evaluated IHES vs CEL, NOS for increased blasts, hypercellularity, abnormal megakaryocytes, dyserythropoiesis and dysgranulopoiesis, markedly elevated M:E ratio and fibrosis, and abnormal eos
• Conclusion: Bone marrow morphology serves as a strong discriminator between CEL, NOS and reactive IHES
Genetics:
Rearrangements of PDGFRA, PDGFRB, FGFR1, BCR-ABL1 must be excluded
- occasionally pts have JAK2 mutation
- X-linked polymorphism analysis of PGK or HUMARA genes in females can be used to demonstrate clonality sometimes
- STAT5B mutations by NGS has been associated with eosinophilia in lymphoid and (rarely) in myeloid neoplasms
Survival is variable (~80% at 5 years)
- S-megaly, inc blasts, dysplasia, and cytogenetic abnormalities have poor prognosis

Myeloproliferative Neoplasm, Unclassifiable
_____________________________________
Useful in pts with definite features of MPN, but do not meet criteria of a specific MPN.
- may be as much as 15% of all MPN cases
Usually fall into one of these categories:
1.) early stage of PV, PMF, or ET, where features are not yet fully developed
2.) advanced stage MPN where fibrosis or other features obscure the real disease
3.) pts c MPN where a neoplasm obscures the underlying disorder
Must consider possibility that dz is not an MPN
- do not just use as trashcan for inadequate sampling or recent cytotoxic tx
Dx / DDx / Genetics
May be difficult to differentiate between early stages of ET, PMF and PV
- PB findings in these can be highly variable, and BM shows hypercellularity with proliferation in all lineages
Late stage disease of MPNs will show lots of fibrosis and exact dx may be impossible without previous labs or bmbx
- inc Mgkcs will help to differentiate from CML in these cases
- should be cautious for MDS or AML progression
MPN is excluded (as usual) if BCR-ABL1, Ph chromosome, or PDGFRA positive and JAK2 may be the only positive genetic finding
_____________________________________
Px is similar to the stage of whichever dz that eventually evolves, with highly advanced dz states having a worse px
Mastocytosis
_____________________________________
Mast cell proliferation that accumulates in 1+ organ systems
- NOTE: mastocytosis is no longer considered an MPN
Dx'ing the subtype depends on location of the dz
clumps of 15+ strangely appearing ( usually spindly w ugly nuclei) mast cells which may be found in various organs (in BM for SM)
-these clumps are also rarely seen in pts in reactive states, tx'd w stem cell factor, or in mast cell leukemia... must differentiate
- these clumps may be arranged in bands, often have dense central mastocytic core

Cutaneous Mastocytosis (CM)
_____________________________________
May occur at any age: Cutaneous Mastocytosis (CM) MC in before 6 mo age, Systemic Mastocytosis (SM) is MC in teens
Sx / DDx / Dx
Darier's sign (urticaria upon palpation) in 80%; may cause bullous mastocytosis (blistering) if severe
- Urticaria Pigmentosa = Darier's sign + melanin accumulation
--BM almost always involved in systemic lesions, yet 50% have cutaneous signs
-- having skin lesions assoc w better prognosis
- other diagnostic criteria: one quarter of mast cells look strange on bx of any organ, KIT 816 mutation, mast cells w CD2/25 on bx of any organ, serum tryptase > 20
Giemsa/ toluene blue/ napthol-ASD-chloroacetate esterase (CAE) can detect mast cell granules
-- they do not express MPO
_____________________________________
IHC: (+) tryptase, chymase (more specific, less sensitive), CD117(nonspecific)/ 2/ 25(not on normal mast cells)/ 9/ 33/ 45/ 68
- mast cells lack CD14/15/16 and most B-/T-cell markers
Genes: KIT mutation activates the tyrosine kinase domain, provides resistance against imatinib (a tyrosine kinase inhibitor)
- MC (33%) is Val for Asp at codon 816 (D816V) in peds c CM
3 major variants:
1.) Urticaria Pigmentosa (UP)/maculopapular cutaneous mastocytosis (MPCM)
- MC variant; spindly mast cells fill papillary dermis, extend to reticular dermis
2.) Diffuse cutaneous mastocytosis
- diffusely thick skin c peau d'orange; band-like mast cell infiltrates
3.) solitary mastocytoma
- single lesion that may extend to subdermis, no atypia; exclusive to infants;
- must also not display signs of SM
Systemic Mastocytosis (SM)
_____________________________________
needs either 1 major + 1 minor or 3 minor criteria:
Major: mast cell clumps on bmbx or bx of other non-skin organ
Minor:
1. in bm/non-skin bx > 1/4 mast cells are spindled / atypical
2. 816 KIT activating point mutation found
3. Mast cells expressing CD 2/25
4. Serum tryptase > 20 (must exclude assoc clonal myeloid disorder to use)
BM almost always involved
B ("burden of disease") findings
1. High mast cell burden (>30% BM mast cells in aggregates and serum tryptase >200)
2. Signs of dysplasia or myeloproliferation, although does not meet criteria for an associated hematologic neoplasm (AHN) with normal or only slightly abnormal blood counts
3. Hepatomegaly without impairment of liver function, palpable splenomegaly without hypersplenism or lymphadenopathy on palpation or imaging
C ("cytoreduction-requiring") findings
1. BM dysfunction caused by neoplastic mast cell infiltrates, manifested by >=1 cytopenia
2. Palpable hepatomegaly with impairment of liver function, ascites, or portal hypertension
3. Skeletal involvement with large osteolytic lesions
4. Palpable splenomegaly with hypersplenism
5. Malabsorption with weight loss due to GI mast cell disease
Variants of systemic mastocytosis:
Indolent Systemic Mastocytosis
- meets general criteria for systemic mastocytosis
- No C findings
- No evidence of associated hematologic neoplasm
- Low mast cell burden
- Skin lesions almost always present
Bone marrow mastocytosis
- same as indolent SM, but with BM involvement and no skin lesions
Smoldering systemic mastocytosis
- meets criteria for SM
- >= 2 B findings, no C findings
- No evidence of associated hematologic neoplasm
- High mast cell burden
- Does not meet criteria for mast cell leukemia
Systemic Mastocytosis with an associated hematologic neoplasm
- meets criteria for SM
- meets criteria for an associated heme malignancy (MDS, MPN, AML, etc)
Aggressive systemic mastocytosis
- meets criteria for SM
- >=1 C finding
- Does not meet criteria for mast cell leukemia
- skin lesions usually absent
Mast cell leukemia
- meets criteria for SM
- Bmbx shows diffuse infiltration by atypical immature mast cells
- BM aspirate shows >20% mast cells
- in classic cases, mast cells >10% of PB WBCs (although aleukemic variant is more common)
- skin lesions usually absent
In SM, if mast cell inflitrates are exclusively round (not spindled) and tryptase-positive, may dx tryptase-positive round cell infiltration of BM (TROCI-BM)
- the TROCI-BM cells may be mast cells, basophils, or myeloblasts
- cellularity changes in area around mast cells should be noted bc affects prognosis
-- eosinophilia and fibrosis also common in these areas
DDx: if considering SM and KIT D816V mutations are negative, consider testing for PDGFRA rearrangements (may be MLNsEO with PDGFRA)
Systemic mastocytosis diagnostic criteria

Mast cell sarcoma
_____________________________________
Mast Cell Leukemia: (+) SM dx criteria and >20% mast cells on aspirate smear. Diffuse infiltration of weird mast cells on bmbx (promastocytes or metachromatic blasts); usually w >10% mast cell on peripheral smear
Mast Cell Sarcoma: High-grade cytology, destructive, unifocal collection of mast cells; w/o SM
Extracutaneous mastocytoma: unifocal tumor w low-grade histo; nondestructive. No SM or skin lesions. IHC: (+) 9, 33, 45, 68, 117, tryptase, chymase (more specific, less sensitive); (-) 14, 15, 16
Tx / Px
"Findings" used in assessing various degrees of mastocytosis (eg indolent or aggressive):
"B" findings:
1. bmbx w > 30% mast cells and/or serum typtase > 200
2. dysplasia in other lineages, but not enough to make a dx (AHNMD), w normal (or almost normal) blood counts
3. Organomegaly (of liver / spleen / lymph nodes) w/o organ dysfunction
"C" findings:
1. BM related cytopenia in any lineage w/o malignancy in that lineage
2. Hepatomegaly w liver dysfunction
3. Osteolytic lesions and/or pathologic fractures
4. Palpable spleen w hypersplenism
5. Malabsorption w wight loss from GI mast cell infiltrates
Prog: favorable in kids, may regress spontaneously around onset of puberty. Mast cell leukemia pts live only a few months. Those w indolent dz have normal life expectancy.
Myeloid / lymphoid neoplasms with eosinophilia and gene rearrangement

Rare; Mutated pluripotent (lymphoid-myeloid) stem cell; Eosinophilia
PDGFRA: CEL, AML, T-LBL
PDGFRB: CMML with eos, AML
FGFR1: T-LBL with eos, CEL, preB-ALL, AML
Both cytogenetics and molecular genetics should be performed in MPNs, ALs, LBLs with eosinophilia
Large number of chromosome translocations
- Fusion gene encodes abnormal tyrosine kinase
- Tyrosine kinase inhibitors may be effective
- PDGFRA and PDGFRB abnormalities are imatinib (Gleevec) responsive
All these abnormalities come from a tyrosine kinase gene aberrantly expressed (thus respond to tyrosine kinase inhibitors [save FGFR1, which doesn't])
- usually see eosinophilia, but not necessary
- probably derived from pluripotent stem-cells
PDGFRA assoc c chronic eosinophilic leukemia (CEL), and lots of mast cells and sometimes neutrophils
- can present as AML or T-LBL, c assoc eosinophilia
PDGFRB has variable presentation, but usually similar to CMML c eosinophilia
FGFR1 has a lymphomatous presentation (T-LBL c eosinophilia)
PDGFRA rearrangement


FISH for CHIC2 deletion -- Arrows indicate loss of CHIC2 signal
FISH PROBES: FIP1L1(G)/CHIC2(R)/PDGFRA(A)[4q12]
Myeloid neoplasm with eosinophilia and PDGFRA-FIP1L1
FIP1L1-PDGFRA fusion abnormally activated tyrosine kinase therapeutic target of imatinib (Gleevec)

Abnormal eosinophil morphology in peripheral blood from a 49-year-old man (case 142) with PDGFRA rearrangement.Eosinophils show abnormal nuclear hypersegmentation, ring form, uneven granulation, or hypogranulation [6]
Myeloid/lymphoid neoplasms (M/LNEo) with PDGFRA rearrangement
_____________________________________
Rare dz; MC in males (17:1 M:F) in 40's; MC rearrangement is FIP1L1-PDGFRA from 4q12 deletion
Diverse morphologic spectrum usually associated with hypereosinophilia
- Presents as CEL, AML, or T-LBL, or all of these at the same time
- blasts <20%, unless AML present
- BM + PB always involved; most organ systems also involved (spleen usually enlarged, skin GI heart lungs CNS etc also); pts present c fatigue or c sx assoc c organ involvement (S-megaly)
-- serum tryptase elevated, but less so than in mast cell dz
• Extramedullary presentation is common ~50% of cases and it can be the primary site of PDFGRA-rearranged neoplasm
• Eosinophilia may be absent (10-20%)
- B12 is usually elevated (versus normal in systemic mastocytosis)
_____________________________________
Micro: Mature, normal-looking eos seen in PB (few precursors); Charcot-Leyden crystals may be seen in tissue sections
BM hypercellular c inc blasts; may appear similar to Systemic Mastosis c inc CD25+ spindly mast cells; reticulin inc
- 2/3 of SM have CD2/25+ mast cells, whereas PDGFRA usually CD2(-)/CD25(+)
_____________________________________
Genes: Cytogenetically cryptic (>90%)
• Locus 4q12
• Most common fusion partner FIP1L1
• Exquisite sensitivity to tyrosine kinase inhibitor
Nested RT-PCR often necessary to detect abnormalities on cytogenetics (which is usually normal)
- using probe for CHIC2 gene c FISH may be helpful; other fusion genes may be present
If rearrangement of PDGFRA by FISH is negative and suspicion is high use other molecular methods (RNAseq, SNP-CN microarray)
_____________________________________
DDx: Aberrant mast cell proliferations (ie considering systemic mastocytosis) in the absence of a KIT mutation are a helpful feature – ~40% of cases – do not make SM diagnosis
-- therefore, when considering Systemic Mastocytosis and is negative for KIT D816V mutation, test for present of PDGFRA
- systemic mastocytosis is resistant to imatinib
_____________________________________
Tx: Pts very responsive to imatinib (100x more so than in CML), complete hematologic remission usually acheived in almost all patients in 1 month
_____________________________________
Px favorable if pt has no cardiac damage






Examples of a spectrum of the mast cell proliferations (resembling systemic mastocytosis) seen in the myeloid neoplasms with FIP1LI-PDGFRA rearrangement. A, B, Case 225: a bone marrow biopsy specimen from a 27-year-old man showing 100% marrow with diffuse eosinophilic infiltrate and mildly increased scatter mast cells, some in small loose aggregates and spindled morphology as highlighted by mast cell tryptase (B). [6]
Case 87: bone marrow biopsy specimen from a 30-year-old-man showing markedly hypercellular
marrow with sheets of mast cells as highlighted by CD117 (E), with aberrant CD25 coexpression (F) in the background of eosinophilia. The mast cells show aberrant loss/decreased expression of mast cell tryptase (D). [6]
A morphologic spectrum of extramedullary involvement associated with PDGFRA rearrangement. A-C, A paraspinal mass from a 39-year-old man showing a myeloid neoplasm with prominent eosinophilia, areas of necrosis, and Charcot-Leiden crystals (case 212). [6]
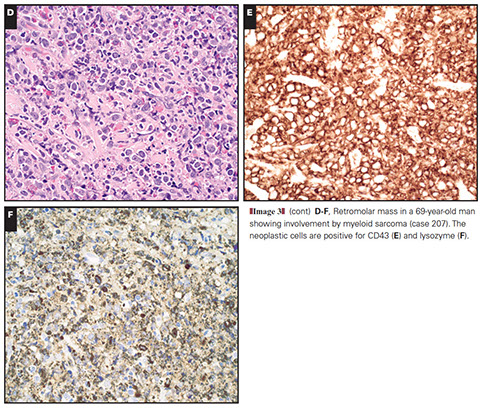
D-F, Retromolar mass in a 69-year-old man showing involvement by myeloid sarcoma (case 207). The neoplastic cells are positive for CD43 (E) and lysozyme (F) [6]
1 - 4
<
>
Myeloid/lymphoid neoplasms with PDGFRB rearrangement
_____________________________________
Rearrangement usually t(5;12)(q31~33;p12) with ETV6-PDGFRB fusion gene formed
Diverse morphologic spectrum usually associated with hypereosinophilia and monocytosis
– CMML, CEL, extramedullary myeloid tumor, rare B-LL
• Usually cytogenetically evident (>95%)
• Locus 5q32
• Diversity of fusion partners: often ETV6
• Exquisite sensitivity to tyrosine kinase inhibitor
Presentation most often similar to CMML or an atypical CML c eosinophilia
- MC in males in 40's
May rarely be cytogenetically cryptic: high index of suspicion, pursue additional testing methods
• Extramedullary presentation may occur/LN with lymphoblastic lymphoma
• Eosinophilia may be absent/mild (~ 20%)
• Atypical mast cell proliferation may serve as a clue
PB shows leukocytosis c anemia and Tbc-penia, with individual WBC lines variably changed
_____________________________________
DDx: BM shows inc mast cells, which can be spindly, with reticulin (possibly) increased; blasts less inc in chronic phase
- mast cells CD2/25 (+), concerning for systemic mastocytosis (SM)
_____________________________________
Genes: ETV6-PDGFRB fusion gene present c t(5;12)(q31~33;p12)
- lots of variants are possible; appropriate FISH studies should be conducted
_____________________________________
Tx: Survival has increased from less than 2 yrs to almost 6 years using imatinib
- treatment with imatinib may be considered in patients with eosinophilia not responding to conventional treatment, assuming that they may have a cryptic PDGFRA or PDGFRB translocation [6]
- cardiac involvement important in determining px
Myeloid neoplasm with eosinophilia and PDGFRB-rearrangement - CMML-like presentation

Myeloid/lymphoid neoplasms with FGFR1 rearrangement
Although derived from pluripotent hematopoietic stem cells, disease may present with precursor or mature cells
- may present as an MPN, AML, or T/B-cell lymphoblastic lymphoma/leukemia (last of these assoc c t(8;13)
- eosinophilia in 70% of cases
Has only moderate male predominance, and presents a little bit younger (in 30's)
- multiple organ systems usually involved, and can present with sx related to organ involvement or more constitutional sx
- considered to have a widely variable clinical presentation
_____________________________________
DDx: similar to diseases with PDGFRA and PDGFRB mutations, may have spindly mast cells positive for CD25, leading one to think of systemic mastocytosis, but should order the PDGFRA, PDGFRB, and FGFR1 mutation analysis if KIT D816V mutation absent
_____________________________________
Genes: Presence of t(8;13)(p11;q12) or variant leading to FGF1 mutation necessary to dx
- trisomy 21 can often be seen
- multiple reported fusion partners
_____________________________________
Px is poor; imatinib has little effect
- interferon has worked in a few pts
MLN with FGFR1 [8]

Myeloid/lymphoid neoplasms with PCM1-JAK2 rearrangement
New provisional entry in 2016 c t(8;9)(p22;p24.1); PCM1-JAK2
- has combo of eosinophilia with left-shifted erythroid predominance, lymph aggs, and often fibrosis in BM
- can rarely present as T- or B-lymphoblastic leukemia which responds to JAK2 inhibition
- originally described as "Ph-negative neutrophilic myelofibrosis"
_____________________________________
Diverse morphologic spectrum usually associated with hypereosinophilia
– MPN, CEL, MDS/MPN, rare lymphoblastic leukemia/lymphoma
Triad [6]:
1) Large immature erythroid islands
2) eosinophilic infiltrates
3) Fibrosis
_____________________________________
Usually cytogenetically evident (>95%)
• Locus 9p24
– Should confirm JAK2 involvement by FISH or alternative modality
• Variants of PCM1-JAK2
– JAK2 with BCR and ETV6 partner genes
• Utility of Ruxolitinib not well known
_____________________________________
Px: disease course and prognosis are highly variable



Characteristic morphologic findings in the myeloid/lymphoid neoplasm with PCM1-JAK2 rearrangement. A-C, Typical bone marrow histopathology features of PCM1-JAK2–rearranged chronic myeloid neoplasm (case 247) showing hypercellularity, large aggregates/nodules of immature erythroblasts (larger area outlined in A and demonstrated by E-cadherin [C]), eosinophilic infiltrate (B, from the smaller area outlined in A), and frequent fibrosis. [6]
Extramedullary involvement is very common in PCM1-JAK2–rearranged neoplasms, often with aggregates of immature erythroblasts and eosinophilia mirroring bone marrow features. Case 255 shows a lymph node with architectural effacement by large sheets of immature
erythroblasts (D and F), as highlighted by CD71 (E and G), consistent with myeloid sarcoma, erythroblastic type. [6]
1 - 2
<
>
Myelodysplasia / myeloproliferation



Chronic MyeloMonocytic Leukemia (CMML)
`
Clonal hematopoietic stem cell disorders
- MDS as well as MPN features
- Leukocytosis, may exceed 100x109/L
- Persistent monocytosis >1x109/L in PB
- Frequent monocytosis in BM
- <20% blasts + promonos in PB and BM
- Dysplasia in one or more cell lines (usually in the granulocytic lineage)
- MDS/MPN in which the main diagnostic feature is peripheral blood monocytosis
• Some cases follow MDS
• Some cases are therapy-related
• Mast cell aggregates (SM) may be seen
Presents as B symptoms appearing as an MPN with leukocytosis and S-megaly
- hallmark is monocytosis in PB (bm often is not really too involved) [8]
80% have Mgkc abnormalities; 30% have fibrosis and 20% have plasmacytoid monocyte nodules on BM
Diagnostic criteria (see table for all 2016 criteria):
CMML-0 has <2% blasts in PB; <5% blsts in BM
CMML-1 has 2-4% blasts PB, 5-9% blasts in BM
CMML-2 has 5-19% blasts in PB, 10-19% blasts BM or if Auer rods are present
JAK2 V617F mutation common (vs other BCR-ABL1 negative MPNs)
Mild eosinophilia may be present, which can mimic PDGFRA/B neoplasms
Myeloproliferative (MP) vs MDS variant
• Myeloproliferative (MP) versus myelodysplastic (MDS) variant
– Dysplastic, WBC <13 x 109/L
– Proliferative, WBC >13 x 109/L
• MP-CMML had shorter overall survival (21 vs 34 months) compared with MDS- CMML.
- the dysplastic and proliferative types have different mutation profiles and prognoses
IHC: CD14 more mature monocytes; lysozyme highlights monocytes and myelomonocytic component
• Flow cytometry better suited
• Monocytic cells: variable expression of CD14, typically co-express CD36/CD64, HLA-DR and CD13 may be diminished, CD33 (bright), CD15 variable, +/- CD56
Alpha napthyl acetate/butyrate esterase alone or in combo c CAE good in assessing monocytic component
- other monocytic antigens are CD13 and CD33
-- CD56 overexpression or CD2 aberrant expression seen
Plasmacytoid dendritic cell (pDC) nodules in CMML
• Present in approx. 20% of cases
• Appears to be clonally related to the CMML
• Not equivalent of BPDCN
Plasmacytoid dendritic cells have (+) CD123/14/43/68/45RA/33(weak)/4
Genes:
Most commonly mutated genes are SRSF2, TET2, and/or ASXL1 (>80%)
- SETBP1, NRAS,KRAS, RUNX1, CBL, EZH2 less common
- ASXL1 mutated CMML have shorter overall survival and shorter AML-free survival
Cytogenetics:
– Abnormal karyotypes seen in 20-30%
– Common: +8, (-Y), -7/del7q,complex
– Several proposed cytogenetic risk categorization schemes
- NPM1 mutation or 11q23 (KMT2A) rearrangements may herald rapid progression to AML and patient should be followed closely
Molecular genetics
– Wide spectrum of abnormalities
– Most common: TET2 (60%), SRSF2 (50%), ASXL1 (40%) and RAS (30%)
• Frameshift and nonsense ASXL1: worse OS
DDx
Reactive monocytoses: malignancy, infection, inflammation, recovering BM post-chemo, G-CSF tx, TB brucellosis, leishmaniasis, endocarditis
Neoplastic: CML, AML, MDS with monocytic progression, MPN with monocytic progression, JMML
Diagnostic pitfalls
• Monocytosis as a component of a reactive phenomenon – infection, inflammatory conditions such as collagen vascular disease, chronic neutropenia, splenectomy, hemolytic anemia, immune thrombocytopenic purpura
• Evolution of CMML-like features in known MPN or MDS. Do not reclassify the disease
• Monocyte morphology: Monocytes vs. abnormal monocytes vs promonocytes (a.k.a. blast equivalents)
Monocytosis in classic MPN
• Do not reclassify disease
• Sign of disease progression
• PMF with monocytosis shows higher JAK2 V617F VAF than CMML (42% vs 17%)
- Must do a BM to prove that the disease is not AML with mature circulating monocytes [7]
Px:
Average survival is 20-40 months
- AML occurs in up to 30%
Blast count is most important prognostic indicator







CMML diagnostic criteria
CMML diagnostic criteria
CMML, PB
CMML, BM
CMML, monocyte morphology, from Dr. Reichard lecture, LASOP, 1/18/2020
CMML, monocyte morphology. Monoblasts and promonocytes have fine nuclear chromatin (maybe the most important feature. Monoblasts have high N:C ratio. Promonocytes have delicate nuclear folding. [8]
CMML, flow
1 - 7
<
>



CMML PDC nodule, ddx for myeloid sarcoma
CMML, PDC nodule, IHC
CMML, PDC nodule IHC
1 - 3
<
>
atypical Chronic Myeloid Leukemia (aCML) - BCR-abl1 negative
By definition, chronic myeloid neoplasm with myeloproliferative and myelodysplastic features concurrently
• Rare; predominantly adult disease
• Manifests predominantly by involvement of the granulocytic lineage characterized by dysgranulopoiesis
• Largely a diagnosis of exclusion
Dysplasia and prolif of neutrophil lineage c leukocytosis from excessive release of neutrophil precursors
Clonal hematopioetic stem cell disorder
- MDS as well as MPN features
- Leukocytosis with left-shifted neutrophils
- Dysplastic neutrophils
May be multilineage dysplasia
- BCR/ABL1, PDGFRA, PDGFRB negative
Variety of cytogenetic and molecular genetic abnormalities
- JAK2 mutations reported
-NRAS and KRAS mutations in ~30%
Seen in older pts c sx from anemia, tcpenia and Smegaly
Diagnostic Criteria
• Peripheral blood leukocytosis >13 x 109/L with neutrophil precursors constituting >10% of leukocytes
• Dysgranulopoiesis
• No or minimal basophilia
• No or minimal monocytosis
• Hypercellular bone marrow with granulocytic proliferation and dysplasia; +/-dysplasia in the erythroid and megakaryocytic lineages
• <20% blasts in the blood in bone marrow
• No evidence of an alternative WHO neoplasm
Genes:
No specific recurring genetic abnormalities
– Trisomy 8, deletion 20q, monosomy 5 seen in a variety of myeloid malignancies
• Driver mutations of MPN (JAK2, CALR and MPL) are typically absent
– 5% of aCML have JAK2
• <10% with CSF3R mutation (mainly CNL)
MC mutations are SETBP1 and ETNK1
- rarely (<1/10) has CSF3R (seen in CNL)
- the driver mutations (JAK2, CALR, and MPL) are typically absent in aCML
Mutations and Frequency
– SETBP1 10-48%
– ETNK1 10%
– NRAS 10-30%
– CSF3R <10% (mostly in CNL)
DDx: Reactive (eg infection, stress, growth factor, etc)
• Chronic neutrophilic leukemia (CNL)
• CNL-like proliferation (underlying plasma cell proliferative disorder/myeloma, solid tumor)
- CML (BCR-ABL1 present)
- CMML (>10% monocytes in blood)
- Genetically-defined eosinophilias (blood eosinophilias, PDGFRA, PDGFRB, FGFR1, PCM-JAK2
Poor prognosis
- survival up to 30 months
-BMT may help survival
Up to 40% evolve into AML, remainder die of BM failure

aCML dysgranulopoiesis in PB

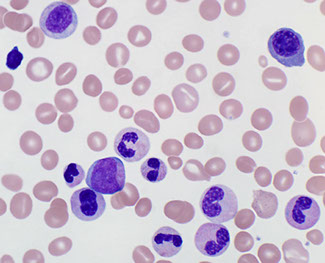
aCML granulocytic dominance

aCML hypercellular c granulocytic proliferation

Juvenile MyeloMonocytic Leukemia (JMML)
Childhood disorder (male predominance) with granulocytic and monocytic proliferation with low blast counts and characteristic RAS/MAPK mutations, usually minimal dysplasia
- assoc c NF1 (10-20% of cases; thus not a criterion)
- kids c Noonan syndrome get a JMML-like dz that resolves spontaneously
- Blasts + Promonos <20% in PB and BM
Present c S-megaly, enlarged tonsils, B symtoms, inc HgF, can have skin and spleen infiltrates
Diagnostic criteria:
I. Clinical + hematologic features (need all 4)
1.) Monocytosis in PB
2.) (Blasts + promyelocytes) <20% in PB and BM; (will never see Auer rods)
3) Splenomegaly
4.) No Ph chr or BCR-ABL1 fusion gene
II. Genetic findings (1 sufficient)
- Somatic PTPN11 or KRAS or NRAS
- Clinical diagnosis of NF or NF mutation
- Germline CBL mutation and LOH of CBL
III For pts w/o genetic features, besides the clinical and hematologic features under I, must have:
- monosomy 7 or any other chromosomal abnormality, or at least 2 of the following:
- Inc HbF; Immature granulocytes in PB; GM-CSF hypersensitivity of myeloid progenitor cells in vitro; hyperphosphorylation of STAT5
PB is most important in dx'ing; see anemia, inc leukos and Tbcpenia
- napthyl acetate/butyrate acetase c NAE good for viz monocytes
Can mimic infx (Ebstein-Barr, CMV, herpes)
~65% have normal karyotype, ~25% monosomy 7
- BCR/ABL1 negative
- RAS and MAPK pathway mutations are characteristic
- PTPN11 gene mutations, SHP-2 phosphatase
- RAS gene mutations
- NF1 gene inactivation
Increased cellular sensitivity to GM-CSF
Hematopoietic stem cell transplantation
Monosomy 7 in 25%; 65% have normal karyotype
DDx: Leukemoid reaction, CML (can occur in kiddos, but very rare), kids c Noonan syndrome have PTPN11 mutation encoding SHP2 or in KRAS
Px: Rapidly fatal if left untreated
- need stem cell transplant or die in 1 year
-- rarely transforms to AML


JMML - myeloid predominance with a left-shift
Myelodysplastic /myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T)
aka RARS-T (former name refractory anemia with ring sideroblasts and thrombocytosis)
- New entity in the MDS/MPN category
- has thrombocytosis, refractory anemia, dyserythropoiesis, and ringed sideroblasts which are >15% of erythroid precursors
- Cases that fulfill the diagnostic criteria for MDS with isolated del(5q) or that have t(3;3)(q21.3;q26.2) or inv(3)(q21.3;q26.2) cytogenetic abnormalities and cases with a BCR-ABL 1 fusion gene are excluded
Assoc c SF3B1 (which is assoc c presence of ring siderblasts) and is usually assoc c comutated JAK2 V617F or less freq c CALR, or MPL genes
Diagnostic Criteria
• Anemia associated with erythroid lineage dysplasia, with or without multilineage dysplasia; >15% ring sideroblasts, need >15% RSs even if SF3B1 mutation present
- < 1% blasts in the peripheral blood and < 5% blasts in the bone marrow (no excess blasts)
• Persistent thrombocytosis, with platelet count >450 x 109/L
• No history of MPN, MDS (except myelodysplastic syndrome with ring sideroblasts - only exception where you can change the previous diagnosis of an MDS!!!), or other MDS/MPN
- SF3B1 mutation (in 90%) or, in the absence of SF3B1 mutation, no history of recent cytotoxic or growth factor therapy that could explain the myelodysplastic/ myeloproliferative features
- JAK2 mutation in 60%
• Absence of BCR-ABL1 fusion; no rearrangement of PDGFRA, PDGFRB or FGFR1; no PCM1-JAK2 and no t(3;3)(q21.3;q26.2), inv(3)(q21.3q26.2), or del(5q)
Genes: Cytogenetic abnormalities uncommon
• SF3B1 mutation seen in >70% of cases
• SF3B1 often (50-60%) seen concurrently with an MPN driver gene mutation (eg, JAK2, MPL, CALR) (majority are CALR)
• Even though these mutations are not needed for the diagnosis, may be helpful to support if on the fence (MDS vs ET)
Px: Much worse than ET
• Better if SF3B1 and/or JAK2 mutated
MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T)

MDS/MPN-RS-T, Hypercellular BM; characteristic megakaryocyte morphology

MDS/MPN-RS-T px


Myelodysplastic / myeloproliferative neoplasm, unclassifiable
Usually due to:
1. Thrombocytosis + anemia and erythroid dysplasia, without ring sideroblasts
2. Thrombocytosis + ring sideroblasts, with excess blasts at initial diagnosis
3. Isolated del(5q) + thrombocytosis, with excess blasts at initial diagnosis
4. Neutrophilia lacking granulocytic dysplasia + dysplasia in another lineage
Typical example of MDS/MPN-U. WBC 10.7, Hgb 11.3, Plt 766. No ring sideroblasts, no monocytosis, normal karyotype, Positive for SETBP1, CSF3R, GATA2, ASXL1 mutations. Can't call atypical CML because no leukocytosis [8]

Myelodysplastic Syndromes
MDS does not progress to MPN, and MPN does not progress to MDS; evolution depends on what the original diagnosis was
- MDS can get fibrosis, JAK2 mutations, and increased megakaryocytes, but you would not say that the MDS progressed to an MPN, just that now you have an MDS with fibrosis [8]
Primary, neoplastic, clonal pluripotent stem cell disorders; karyotype often normal or has abnormalities seen in MDS
- One or more cytopenias in blood (ineffective hematopoiesis)
- Maturation abnormalities in bone marrow (dysplasia/dyspoiesis)
- Progressive cytopenias and BM failure c potential evolution to acute leukemias, previously known as “preleukemias”
- MC in elderly, rare in children, correlates c advancing age, therapy-induced DNA damage and shorter telomeres
- Males > Females
- may be caused by environmental (chemo/radiation, benzene, smoking, agricultural chemicals) or genetic (Shwachmann-Diamond and Diamond-Blackfan syndromes, Fanconi anemia and dyskeratosis congenita) factors
-- should not call (primary?) MDS if pt is on growth factor therapy (?, ie EPO)
Abnormal cells = clones of abnormal stem cell
- c dz progression, abnormal clone predominates
MC mutated genes are SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, EZH2
- >50% have chromosome abnormality at dz
- MC cytogenetic abnormalities: deletions of chromosomes 5 and 7, and trisomy 8
- the number and types of translocations are strongly assoc c outcomes; further genetic events assoc c AML
- TP53 mutation eval recommended in pts c iso(5q) bc assoc c resistance to lenalinomide and assoc c worse px
Clonal Hematopoiesis of Indeterminate Potential (CHIP) is acquired clonal mutations identical to those seen in MDS that can occur in hematopoietic cells of apparently healthy older individuals
-CHIP Appears to be a precursor state to MDS
- Analogous to the relationship of MGUS to myeloma and monoclonal B-lymphocytosis to CLL
- Most patients with CHIP do not develop MDS
- In the current WHO classification, CHIP phenomenon precludes the current use of mutations in isolation to diagnose MDS
Clonal Cytopenia of Undetermined Significance (CCUS)
- Specific mutation patterns and high mutant allele frequency may confer higher risk of MDS
- Malcovati L et al. Blood 2017;129:3371
- Studied a cohort of unexplained cytopenia
- Mutant allele fraction ≥10%, PPV 0.86; >2 mutations, PPV 0.88 for a diagnosi of a myeloid neoplasm
- Spliceosome gene mutations and comutations of TET2, DNMT3A or ASXL1 mutation had PPV of 0.86-1.00
Intrinsic defects in cellular maturation and function associated with both MDS and AML
- Ineffective hematopoiesis secondary to increased apoptosis
-- Increased TNF-alpha in bone marrow mediates apoptosis
- Abnormal levels of IL-1, IL-6, IL-8 in serum and bone marrow
Decreased apoptosis associated with progression to acute leukemia
Primary MDS
Don’t know exact etiologies; De novo
Genetic damage accumulates
- Oncogene activation
- Tumor suppressor gene inactivation
- Impaired apoptosis later in disease course
- Telomerase activation
- usually in older adults
Secondary MDS
Therapy-related MDS
- Chemotherapy (e.g. alkylating agents)
- Radiation therapy
Environmental mutagens/toxins (e.g. benzene)
- can be seen in Children
- Constitutional chromosomal disorders (e.g. Down Syndrome)
- Immunodeficiency disorders (e.g. Wiskott-Aldrich Syndrome)
Sx
- Fatigue and weakness due to anemia
- Less common sx: Infx + fever 2/2 neutropenia
- Bleeding due to thrombocytopenia
Many persons asymptomatic at diagnosis
Infection and hemorrhage are leading causes of death
Organomegaly is uncommon
Cytopenias (anemia, neutropenia, tbcpenia)
- Isolated cytopenia - 35%; Bicytopenia - 30%
- Pancytopenia - 20%
Dysplasia of one or all cell lines typical
- Dyserythropoiesis is the hallmark of MDS
Functional abnormalities are common
- Worse cytopenias = worse prognosis
Uni-/bicytopenia c UNI-lineage dysplasia --> RCUD (RA/RN/RT)
" " BI-lineage dysplasia (or more; +/- ringed sideroblasts) --> RCMD
Pancytopenia c UNI-lineage dysplasia --> MDS-U
Low power morphologic abnormalities in MDS
- hypercellular marrow (80% of cases)
- disorganization of hematopoiesis
-- immature myeloid elements occur away from bone trabeculae
-- erythroid elements fail to form well-defined clusters (no erythroid islands!!!)
- megakaryocytes may cluster
Dysplastia is not really reproducible: Font P Ann Hematol 2013;92:19, Parmentier S Hematologica 2012; 97:723, Matsuda A Leukemia 2007: 21;678, Della Porta MG Leukkemia 2014;29;66
Dys-ERYTHRO-poiesis = multinuclear, budding, megaloblastic, karyorhectic nuclei; PAS(+)/vacuoled cytoplasm
- anemia, macrocytes, oval macrocytes, dimorphism, basophilic stippling, nucleated RBCs, Howell-Jolly bodies, sideroblasts, anisocytosis, poikilocytosis, reticulocytosis
Dys-GRANULO-poiesis = small, hypo-/hyperlobated nuclei with hypo- or pseudo Chediak-Higashi granules
Neutropenia, hypogranulation, abnormal granulation, left-shift, nuclear hyposegmentation (pseudo-Pelger-Huet, ring nuclei), Auer rods, monocytosis
- presence of Auer rods automatically upgrades to high-grade RAEB-2 or CMML-2, and has poor px, but is not automatically considered AML
Dys-MEGAKC-poiesis = hypo-/non-lobate or multiple, separated nuclei and overall small size
Thrombocytopenia to thrombocytosis, giant forms, hypogranulation, micromegakaryocytes, functional abnormalities (abnormal aggregometry)
BM findings
Usually hypercellular marrow for age with erythroid hyperplasia, but may see normocellular or hypocellular marrow
Generally increased number of blasts
Generally trilineage dysplasia (dyshematopoiesis)
- Abnormal localization of immature myeloid precursors (ALIP)
Cytogenetic abnormalities seen in 50% of MDS
- isolated del(5q) assoc c hypo-/non-lobated Mgkcs, macrocytic anemia, inc platelets, and good px, esp in women
- 17p loss assoc c pseudo PH cells, small vacuolated neuts, TP53 mutation, poor px, and assoc c tx-related MDS
- complex karyotypes assoc c poor px
- isolated del(20q) usually involes erythroid and Mgkcs and good px
- chr 7 changes assoc c poor px
Intl. Prognostic Scoring System (IPSS) categorizes % blasts, karyotype, and # cell lines c cytopenia and gives px
- blast % carries the strongest info about px (?)
Lab findings
Serum iron - normal or increased
TIBC - normal or decreased
Serum vitamin B12 and folate - normal or increased
- Decreased total numbers of T-cells and T-cell response to mitogens
- Normal numbers of B-cells, but serum hypergammaglobulinemia and circulating immune complexes
Decreased granulocytic oxidative metabolism
Impared granulocytic chemotaxis
Avoid MDS diagnosis if potential secondary cause
- drugs/toxins
-- recent (<6 mo) chemotherapy or radiation therapy
- arsenic/heavy metal toxin
- alcoholism
- vit B12/folate dificiency, copper deficiency
- Infections (AIDS)
- Neoplasms
-- involving marrow (esp myeloma, LGL, HCL)
-- Beware of making the DX in young patients!!!!!!!Q!
Flow in MDS
- Flow is important to eval for lymphomas that can prsent with cytopenia mimicking MDS
- abnormal flow patterns predict MDS with good sensitivity and specifiicity
---- abnormalities in blasts and maturing elements
- WHO 2017 and ELN guidelines do not permit a dx of MDS s9lely based on flow cytometry


Dysmegakaryopoiesis

Dyserythropoiesis

Dysgranulopoisis

Dysmegakaryopoisis




Meg dysplasia

Granulocytic dysplasia

Erythroid dysplasia [3].

Flow MDS

Refractory Anemia (BM)

MDS-defining cytogenetics, 50% with normal karyotype
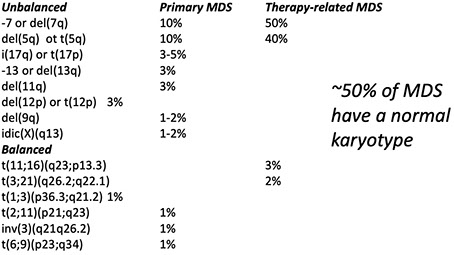

MDS vs AIHA, low power clue, MDS is more disorganized, AIHA shows increased erythroid precursors, compensating correctly for the anemia
MDS genetics [3]. A sig portion of healthy pts have somatic MDS-type mutations in hematopoietic cells (DVMT3A, TET2, TP53, SF3B1)
- nc incidence w age
- asoc c inc risk of malig but many never get MDS, called CHIP or CCUS if pt cytopenic

CHIP and anemia are frequent in elderly pts, while MDS is rare. Ma X Am J Medicine 2012;125: S2, Rollinson DE Blood 2008; 112:45-52, Jaiswal S NEJM 2014;371:2488, Steensma D Blood 2016; 126:9

MDS diagnosis [3], must rigorously exclude secondary causes if diagnosing by morphology, diagnosing MDS in a vacuum is reckless

MDS diagnosis and IPSS-R [3]

MDS risk stratification: both IPSS-R and WHO provide complimentary and clinically relevant data. Malcovati L et al. J Clin Oncol 2005; 237594, Greenburg et al Blood 2012; 120:2454; clinicians may tello pts both WHO category and IPSS risk score


Cytogenetics in MDS, stratifying risk [3]. Schanz et al. J clin oncol 2012 30
Geen px mds [3, listed in dec freq

Figure 6. Hematopathology alphabet soup: a quick comparison. With CHIP, ICUS, CCUS, and MDS. From Albert C. Integrating NGS into the cytopenia workup. CAP Today; May 2022.

MDS diagnostic algorithm tree. From Albert C. Integrating NGS into the cytopenia workup. CAP Today; May 2022.

MDS with single lineage dysplasia
Dysplasia and cytopenia in 1 of the 3 cell lines, including: Refractory Anemia (RA), Refractory Neutropenia (RN), and Refractory Thrombocytopenia (RT)
- can have cytopenia in 2 cell lines, but can only have dysplasia in 1
-- Pancytopenia c 1 cell line dysplasia termed MDS-U
RCUD comprises up to 20% of all MDS cases
- MC RCUD is RA (RN and RT are very rare)
Mean age at dx: 70 years
Sx related to specific cytopenia
Dyserythropoiesis = megaloblastoid maturation, impaired hemoglobinization and and oval macrocytes
- No dysgranulopoiesis or dysmegakaryocytopoiesis
- WBC and Plts are normal or low.
- Mild to severe anemia
- MCV and RDW usually elevated.
Dysplastic NRBCs, sideroblasts and siderocytes can be seen in the PB
- Reticulocyte count may be high or low.
<1% blasts in PB, <5% in BM
Must have >10% of cells in a particular cell line affected to diagnose dysplasia
Must r/o exogenous causes of dysplasia, such as toxins, growth factor tx, viral infx, immunologic / congenital dz, vitamin deficiency / excess (ie zinc supplementation)
If no cytogenetic abnormality present, must wait 6 months to dx
_____________________________________
Px:
Most cases have good long-term survival px, and only a small % progress to AML
- cytogenetic abnormalities are usually those assoc c good px also
-- 90% + of pts live 2 years +, having similar life-expectancies to unaffected population
MDS SLD [3]



Dysmegakaryopoiesis
MDS with ring sideroblasts (MDS-RS)
Usually requires >15% ring sideroblasts; however the diagnosis can be made with >5% in the presence of SF3B1 mutation
- Secondary causes of ring sideroblasts must be excluded.
<1% blasts in PB and <5% in BM
- Megaloblastoid maturation with impaired hemoglobinization.
- Minimal dysgranulopoiesis / megakaryocytopoiesis
SF3B1
- Strong association of MDS-RS with SF3B1 mutation
- Seen in ~70% of MDS-RS cases
- Probable early event in disease development
- SF3B1 is a spliceosome gene
- Favorable clinical outcome
Ring sideroblasts are erythroid precursors with mitochondrial Ferritin accumulation
- most likely a defect in mitochondrial iron accumulation and not a primary defect in heme synthesis
-- probably caused by mitochondrial DNA mutation/deletion (ie Pearson marrow-pancreas syndrome)
Ineffective erythropoiesis caused by abnormal Fe metabolism that results in Fe deposition in erythroid lineage at early stages
Can see evidence of Fe overload in liver and spleen
Sx usually related to anemia, though Fe overload sx can be present
Anemia is usually normo/normo or normo/macro
- no blasts in PB
BM shows increased dysplastic erythoid precursors and (usually) hyperplastic marrow
- often will see hemosiderin-laden macrophages
Must exclude use of alcohol, toxins/drugs (Pb, benzene, zinc, isoniazid), Cu deficiency and congenital siderblastic anemia
Up to 20% have a clonal abnormality, which is usually of a single chr
Avg survival usually more than 5 years
- low % progress to AML

MDS RS [3] - abnormal pearls in ring sideroblasts is actually mitochondria, as a string of pearls, can be caused by alcohol and medications / toxins, or hereditary sideroblastic anemia, needs to encircle at least 1/3 of the nucleus

MDS with multilineage dysplasia
MDS c dysplasia in 2+ cell lineages
- <1% blasts in PB, <5% in BM; no Auer rods
-- greater % blasts in PB or BM +/- Auer rods should be dx'd the appropriate RAEB
- RCMD may have >15% ring sideroblasts; BM is usually hypercellular
- 30-50% have de novo chromosomal abnormalities
-- usually non-specific; 5q-, monosomy 7 or del(7q)
-- Commonly have multiple abnormalities
Mean age at dx >70 yrs c slight male predominance
- Sx related to specific cell line cytopenia
Up to 50% of RCMD can have tri 8, mono 7, del (7q), mono 5, del(5q), del(20q) or complex karyotypes
Falls in the intermediate px category of IPSS, and depends on the amout of cytopenia and dysplasia
- complex karyotypes have worse px, similar to pts c RAEB
Up to 10% progress to AML in 2 years
Overall survival is mean of ~2.5 yrs
MDS MLD [3]

MDS with excess blasts
Same erythroid changes seen in RA or RARS.
Dysplasia and cytopenia of two cell lines.
- More pronounced dysgranulopoiesis
MDS c 5-19% blasts in BM or 2-19% blasts in PB; 2 categories (RAEB-1 or RAEB-2)
- RAEB-1 has 5-9% blasts in BM, 2-4% blasts in PB
- RAEB-2 has 10-19% BM blasts, 5-19% PB blasts; Auer rods automatically qualifies as RAEB-2
RAEB comprises ~40% of MDS cases
Mean age at dx >50 years
Risk factors: envt'l toxins (pesticides, petroleum derivatives, heavy metals), cigarette smoking
Sx assoc c anemia, tbc-penia, neutropenia
All 3 cell lines usually affected, with their own particular dysplastic features (outlined in "MDS overview"); BM usually is hypercellular (but may be normo- or hypocellular)
- erythro- and mgkc-poiesis usually occur in paratrabecular region, which is usually where granulopoietic cells reside
15% of all MDS cases have significant fibrosis (highlighted c reticulin stain) and is coined MDS-F, most of which cases are RAEB-F (if other RAEB criteria apply)
- need to exclude non-neoplastic causes of fibrosis (tx, or HIV); and marrow should not have lots of collagen
-RAEB-F can have overlap c Acute Panmyelosis c Myelofibrosis (APMF), which has abrupt onset fever c bone pain
CD34/117 helps to highlight blasts, which also have asynchronous CD15/11b/65 expression
- 20% of blasts may show CD7 (poor px), and 10% can have CD56
Up to 50% have clonal cytogenetic abnormalities, such as +8, -5, del(5q), -7, del(7q), del(20q) and complex karyotypes
25% of RAEB-1 and 33% of RAEB-2 progress to AML; rest succumb to BM failure
Mean survival for RAEB-1 is ~1 year; RAEB-2 is <1 year
- high PB blast % in RAEB-2 has px similar to AML (~3 mo survival); whereas survival in those with just Auer rods is slightly higher (~1 year)
- (+) CD7 assoc c poor px
MDS EB [3], once you have excess blasts, ring sideroblasts not relevant, and iso del 5q dont matter

Blast counting MDS [3]

Blasts MDS

Blasts! [3]. CD34, even granular forms count. CD117, also stains erythroid precursors but more lightly, and also mast cells darkly, as well as promyelocytes (more useful is CD34 negative). CD61. See megs and 2 micromegs

ALIP [3], clusters of blasts that are CD34+

Myelodysplastic syndrome with excess blasts and erythroid predominance
Previously, erythroleukemia (erythroid/myeloid-type acute erythroid leukemia) were cases of myeloid neoplasms where maturing erythroblasts were >50% of the marrow cells and myeloblasts were >20% of non-erythroid nucleated marrow cells
- now blasts are just classified according to the blast percentage of all cells, regardless of the number of erythroid cells
Genes: TP53, RUNX1, and ASXL1 mutations assoc with poor px in MDS-EB with erythroid predominance
- NPM1 mutations, which are uncommon in this group, may define a subset with a favorable px if given intensive chemo
Myelodysplastic syndrome with excess blasts and fibrosis (MDS-EB-F)
In ~15% of MDS cases, there is grade 2-3 reticulin fibrosis, and this is termed MDS-F, and most are in the EB category and should therefore be called MDS-EB-F
- marrow fibrosis can also be increased in cases of therapy-related myeloid neoplasms, MPNs, lymphoid neoplasms, and reactive conditions (infx and autoimune disorders), which must be ruled out
Micro: megakaryocytes are usually increased and are dysplastic
IHC: aberrant CD7 expression in blasts in 20%, aberrant CD56 expression in 10%
DDx: acute panmyelosis with myelofibrosis (this entity has an abrupt onset with fever and bone pain, and has a higher blast count)
Px: 25% of MDS-EB1 and 30% of MDS-EB2 progress to AML
Myelodysplastic syndrome with isolated del(5q)
-aka Refractory macrocytic anemia with megakaryocytic abnormalities, or “5q- Syndrome”.
- Stable course, often transfusion dependent
- Female preponderance, elderly
PB is similar to RA, 50% have thrombocytosis
BM has numerous small to medium-sized mononuclear or bilobed megakaryocytes with vacuoles.
Updates
No adverse effect on clinical outcome with one additional cytogenetic abnormality except -7 and del(7q)
- The presence of a TP53 mutation is associated with an inferior response in patients treated with lenalidomide
MDS c anemia and no other cell lineage abnormality, that of course has only a del(5q)
- should not have excess blasts in PB or BM and no Auer rods
Early Growth Response 1 (EGR1) and CTNNA1 tumor-suppresor genes in 5q32 possible causes
- A gene that encodes ribosomal proteins, RPS14 gene, is another possibility
Pt usually has severe, macrocytic anemia, and 1/3 have thrombocytosis
BM usually shows hypercellularity and erythroid hyperplasia
5q gene normally helps to differentiate between the 3 cell lines, but not the lymphoid cell line
Survival is >10 years and <10% progress to AML
- other genetic abnormalities or excess blasts should be reclassified and have poorer px
Thalidomide analog lenalidomide tx has better px; and transfusion independence (?) achieved in 2/3 of pts and is related to suppression of the abnormal gene
MDS c isolated 5q del

MDS isolated del 5q [3]

Myelodysplastic syndrome, unclassifiable
Type of MDS that doesn't fit other MDS categories, usually in following situations:
1.) 1% blasts in PB, <5% BM blasts
- technically doesn't reach MDS-EB1
2.) Pancytopenia with unilineage dysplasia (MDS-SLD only allows mono-/bicytopenia)
3.) Clonal cytogenetics abnormality + No dysplasia
MDS-U should be closely followed for evolution into a more specific type of MDS
Unknown what % transform to AML
Cases similar to RCUD or RCMD which have 1% blasts in PB have px worse than RCUD/RCMD but better than RAEB
MDS-U [3]

Childhood myelodysplastic syndrome
MDS is very uncommon is pts <14 yo, and must be distinguished from other causes that can dause dysplasia (tx or other conditions), and ring sideroblasts and isolated del(5q) are especially rare
- ~1/4 cases of childhood MDS are in pts c Down syndrome, though now this dx is questionable and may be considered a distinct entity
Children MC present c neutro- and tbc-penia, vs anemia usually seen in adults, and hypo-cellularity (vs hypercellularity in adults)
- distinction of RAEB in kids, c same criteria as in adults, should be distinguished, although its significance not yet known, and these cases should be closely followed for progression to AML
If any of the cytogenetic abnormalities assoc c AML are found in BM, best to dx AML regardless of blast counts
Mono 7 is MC genetic abnormality, though there have not been many cases to be certain
- cytogenetic abnormalities do not affect morphology
Karyotype is most important factor in determining disease progression
- monosomy 7 has high probability of disease progression
- trisomy 8 assoc c long and stable course of dz
HSCT is only tx that may provide cure for pts c mono 7 or other complex karyotypes
Refractory cytopenia of childhood (RCC)
MDS c persistent cytopenia and <5% blasts in BM, <2% in PB
- majority of pts show significantly hypocellular BM
MCC MDS in childhood (~50% of all cases)
- spleen, liver and LNs are not sites of primary involvement
MC presenting sx are malaise, bleeding, fever and infx, though 20% have no sx
Anisopoikilocytosis and macrocytosis of RBCs and platelets common on PB
- PH cells and hypogranularity of neutros seen
- further dysplastic changes outlined in "MDS Summary" above
Most bmbx have hypocellular cores, making it hard to differentiate from BM failure disorders, and myelodysplasia due to secondary causes (infx, vitamin deficiency, metabolic disorders)
- pts c Down syndrome related neoplasms are excluded
Aplastic anemia, inherited BM failure diseases and PNH must be excluded
- precursor cells in aplastic anemia in children are not dysplastic!
- congenital diseases may have overlap c RCC (ie Fanconi anemia, dyskeratosis congenita, Scwachman-Diamond syndrome, amegakaryocytic thrombocytopenia, pancytopenia c radioulnar synostosis)
Most RCC cases have no cytogenetic abnormality
Mimickers of MDS
Pseudo–Pelger-Huët abnormalities
Etiology: number of medications
Neutrophils demonstrate hyposegmentation with abnormally clumped chromatin; Dohle bodies also
Cytologic changes typically abate after removal of the offending agent
Be careful to not over interpret as myelodysplasia!
Copper Deficiency
Cytopenias
Bone marrow findings
Variable cellularity
May be hypocellular, normocellular, or hypercellular
Possible erythroid predominance
Cytologic
Distinctive cytoplasmic vacuoles in granulocytic and erythroid precursors
Megakaryocytes may show some slight atypia
May be some subtle nuclear-cytoplasmic dyssynchrony in the erythroid or granulocytic precursors
No overt dysplastic features
Blasts not increased
Ring sideroblasts are common
Main neoplastic differential diagnosis is with low-grade MDS
Should not see vacuolated granulocytic precursors in MDS
Tips: need clear-cut dysplasia in MDS fulfilling WHO criteria or a second timepoint on the curve after exclusion of non-neoplastic mimickers
Differential of non-malignant dyspoiesis
Avoid MDS diagnosis if potential secondary cause
-- Beware of making the diagnosis in young patients!!!!!!!! Vitamin/micro nutrient deficiencies: copper (ring sideroblasts, vacuoles), vit B12/folate dificiency Infections: HIV (multilineage dyspoiesis)
Toxins/drugs: EtOH (acanthocytosis, ring sideroblasts), MMF (pelgeroid neutrophils), recent (<6 mo) chemotherapy or radiation therapy, arsenic/heavy metal toxin
Autoimmune/rheumatologic Congenital: dyserythropoiesis due to increased RBC fragility, baseline dyspoiesis and bone marrow failure syndromes
Neoplasms: involving marrow (esp myeloma, LGL, HCL)

Copper deficiency


Post-chemo dyspoiesis [4]

Myeloid neoplasms with germline predisposition

Although MDS and acute leukemia are sporadic dzs, a subgroup is assoc c germ line mutations and is familial
- the presence of a specific underlying genetic defect or predisposition syndrome should be noted as part of the dx
Broken down into 3 general groups:
1) Myeloid neoplasms with germline predisposition without a pre-existing disorder or organ dysfunction
2) Myeloid neoplasms with germline predisposition and pre-existing platelet disorders
3) Myeloid neoplasms with germline predisposition and other organ dysfunction

1) Myeloid neoplasms with germline predisposition without a pre-existing disorder or organ dysfunction
- has two groups:
a) AML with germline CEBPA mutations
b) Myeloid neoplasms with germline DDX41 mutations
a) Acute myeloid leukaemia with germline CEBPA mutation
- Inheritance of a single copy of mutated CEBPA, which encodes a granulocyte differentiation factor on cr 19q13.1
- appears to have near complete penetrance for the development of AML, though the prevalence is unknown
- there are a significant amount of cases with biallelic mutations, where one of the mutations seems germline and the other somatic
Px: favorable overall, though multiple relapses reported
b) Myeloid neoplasms with germline DDX41 mutation
- inherited mutation on cr 5 encoding the DEAD box RNA helicase DDX41
- similar to CEBPA mutations, one of the mutations may be inherited and the other somatic
Px: Poor
2) Myeloid neoplasms with germline predisposition and pre-existing platelet disorders
- includes 3 categories:
a) Myeloid neoplasms with germline RUNX1 mutation
b) Myeloid neoplasms with germline ANKRD26 mutation
c) Myeloid neoplasms with germline ETV6 mutation
a) Myeloid neoplasms with germline RUNX1 mutation
- autosomal dominant syndrome with abnormal platelet number and function, with increased risk of developing AML at a young age
- RUNX1 encodes a subunit of the core binding transcription factor that regulates expression of genes essential for hematopoiesis on 21q22
Sx: variable, most pts with mild to moderate bleeding tendency
- median age of onset of MDS/AML is 33 years
-- may also prediscopse to CMML, T-ALL, and B-cell neoplasms
- anticipation appears to occur in the pedigrees
b) Myeloid neoplasms with germline ANKRD26 mutation
- Autosomal dominant disorder, Thrombocytopenia 2, with moderate thrombocytopenia and increased risk of MDS/AML
- assembly of RUNX1 and FLI1 on the ANKRD26 promoted is disrupted, causing increased gene transcription and signaling through the MPL pathway, which in turn causes impaired proplatelet formation by megakaryocytes
Most patients have GP 1a and alpha-granule deficiency, whereas in vitro platelet aggregation studies are often normal
Germline Predisposition Syndrome
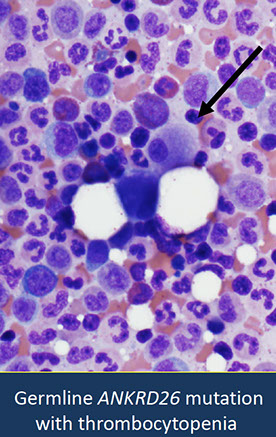
c) Myeloid neoplasms with germline ETV6 mutation
- Thrombocytopenia 5 (germline ETV6 mutations) is a recently described disorder with AD familial thrombocytopenia and hematologic neoplasms
3) Myeloid neoplasms with germline predisposition associated with other organ dysfunction
- includes 5 categories:
a) Myeloid neoplasms with germline GATA2 mutation
b) Myeloid neoplasms associated with bone marrow failure syndromes
c) Myeloid neoplasms associated with telomere biology disorders
d) JMML associated with NF, Noonan syndrome, or Noonan syndrome-like disorders
e) Myeloid neoplasms associated with Down syndrome
a) Myeloid neoplasms with germline GATA2 mutation
- originally identified as four separate syndromes:
1) MonoMAC syndrome: has monocytopenia and non-tuberculous mycobacterial infection
2) Dendritic cell, monocyte, B- and NK-lymphoid (DMCL) deficiency with vulnerability to viral infections
3) Familial MDS/AML
4) Emberger syndrome: with primary lymphedema, warts, and a predisposition to MDS/AML
Has also been seen with congenital neutropenia and aplastic anemia
GATA2 is a zinc-finger transcription factor regulating hematopoiesis, autoimmunity, and inflammatory and developmental processes
- can be seen in a fairly large proportion of kids with MDS
Micro: BM hypocellularity and multilineage dysplasia such as micromegs and megs with separated nuclear lobes, as well as increased reticulin fibrosis
- by flow, there is abnormal granulocyte maturation, monocytopenia, and decreased numbers of BM NK and B-cells
Genes: also associated with monosomy 7 and trisomy 8
Px: poor, although number of cases is limited
MDS with germline GATA2 mutation: typically in a patient with history of immunodeficiency showing monocytopenia, hypocellular BM, and micromegs

Another micromeg in a patient with germline GATA2 mutation

b-c) Myeloid neoplasms with germline predisposition associated with inherited bone failure syndromes and telomere biology disorders
- Include diseases such as Fanconi anemia, Shwachman-Diamond syndrome, Diamond-Blackfan anemia, and severe congenital neutropenia
-- may not manifest until adulthood
Acute myeloid leukemia (AML) and related precursor lesions
Acute Myeloid Leukemia (AML) with recurrent genetic abnormalities
Not really a single dz, but is complex and heterogeneous
- AML is a proliferation >20% myeloid precursors (blasts) in PB or BM
- blast count <20% may be considered AML if the typical translocations are present (though not entirely clear-cut)
- these translocations produce chimeric proteins that influence marrow production of cells
Genetic testing required on all or most AML:
FLT3-ITD, NPM1, CEBPA, RUNX1
Others: IDH1, IDH2, TET2, WT1, DNMT3A, TP53, others
- KIT for core-binding bactor leukemias, PML-RARA upfront if APL is suspected
4 MC genetic alterations are
(1) t(8:21)(q22:q22); RUNX1-RUNX1T1
(2) inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11
(3) t(15;17)(q22;q12); PML-RARA
(4) t(9;11)(p22;q23); MLLT3-KMT2A
Discussed in more detail in the box are these and others common to AML
Start with large dose induction chemo to try and send cancer into remission
- give ATRA for acute promyelocytic leukemia
BMT may offer cure for some pts
Px overall is pretty poor, most (80%) do not make it past 3 years
- refer to cytogenetic tests to more accurately establish px
- FLT3 mutations assoc c worse px, while CEBPA and NPM1 assoc c good px in AML c normal karyotype
FAB subtypes of AML
M0 - Undifferentiated AML
M1 - AML with minimal maturation
M2 - AML with maturation
M3 - Acute promyelocytic leukemia (APL)
M4 - Acute myelomonocytic leukemia
M5 - Acute monocytic leukemia
M6 - Acute erythroid leukemia
M7 - Acute megakaryoblastic leukemia
-crop-u29698.jpg?crc=328485397)
Blast with Auer rod

Blasts in AML
From Dr. Reichard lecture at City of Hope, Jan 18, 2020, LASOP

Grimwade, D. et al. Blood. 1998;92:2322-2333
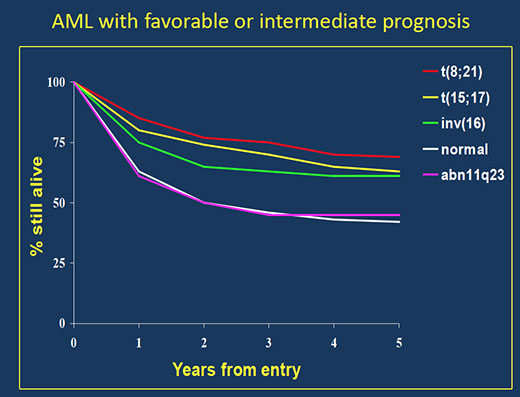




Non-blast features in AML

LeukemiaNet Prognostic Genetic Categories [4]

AML survival among European LeukemiaNet risk categories [4] [Rollig C et al. JCO 2011;29:2758-65]
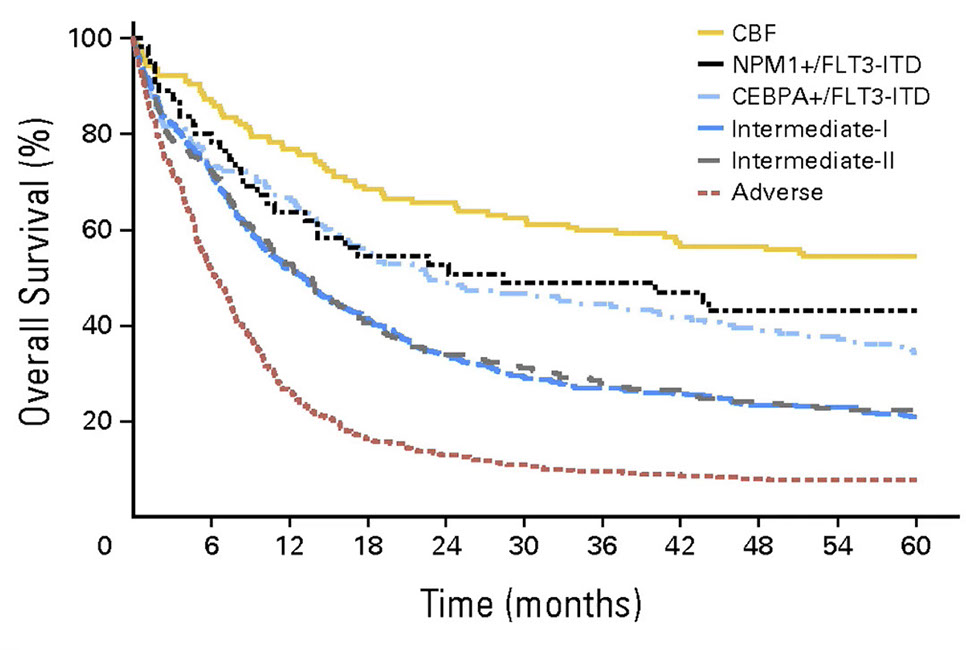
FLT3 in AML (not a subtype)
FLT3 is a receptor tyrosine kinase
- FLT3 mutations 30%; 25% ITD, 7% TKD
- Both result in constitutive activity
FLT3-ITDhigh is a driver mutation that confers a poor prognosis with an increased risk of relapse and shorter overall survival (OS) compared with patients without the mutation
FLT3 testing be performed at diagnosis in all patients to identify those who may benefit from targeted treatment options
• FLT3 ITD mutations
– 20% of AML cases
– Poor prognosis and resistance to standard therapy
• FLT3 TKD mutations
– Prognostic relevance is controversial
TP53 in AML
Frequency: 5-15%, increases with age (< 60 years: ~5-10%; > 60 years: ~10-20%)
- Associated with complex karyotype, monosomal karyotype, –5/5q–, –7/7q–, abn(17p)
- Associated with very poor outcome
- TP53 mutation and complex karyotype provide independent prognostic information, with the combination of both lesions having dismal outcome
ASXL1 in AML
- Early event in leukemogenesis
- 3-19% mutation frequency
- Frequency increases with age (< 60 years: 5-10%; > 60 years: 10-18%)
- Mutations appear to be more frequently associated with AML with myelodysplasia-related changes and a worse overall survival
- ASXL1/RUNX1 and ASXL1/SRSF2 mutations particularly poor outcome
AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1
- Seen in approximately 5% of AML cases
- More frequent in younger patients
- may present as myeloid sarcoma
- Germline mutations should be evaluated
- Diagnosis restricted to de novo cases (exclude history of MDS or therapy)
- Cases with MDS-related cytogenetic abnormalities should be classified as AML-MRC
- May be associated with other mutations
- KMT2A-PTD, IDH1, IDH2, ASXL1
- Rare CEBPA or NPM1
- Cases with RUNX1 and NPM1 mutations should be classified as AML with mutated NPM1
- Cases with RUNX1 and biallelic CEBPA mutations should be classified as AML with biallelic mutations of CEBPA
Micro: blasts typically have basophilic cytoplasm and azurophilic granules; frequently have Auer rods
- dysplasia is commonly seen only in the granulocytic lineage, the erythroids and megs look ok
IHC: (+) CD34, HLA-DR, MPO, CD13, CD33 (weak)
- can show neutrophilic differentiation with CD15+ or CD65+ blasts, in which case may express lymphoid markers CD19, PAX5, and CD79a, and may be weakly positive for TdT
- CD56 associated with worse prognosis (may be associated with KIT mutations)
Genes: RUNX1 encodes the alpha subunit of CBF, which is essential for hematopoiesis
- KIT mutations occur in 20-30% of cases
Px
- Good response to therapy
Blasts in t(8;21)(q22;q22) - see perinuclear clearing, azurophilic (large pink) granules and Auer rods [4]

Acute myeloid leukemia with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11
- shows monocytic and granulocytic differentiation and characteristically an abnormal eosinophil component in the BM
- seen in 5-8% of younger pts with AML, less common in older adults
- myeloid sarcoma may be present at dx
- WBC count significantly higher in pts with inv(16) or t(16;16) than in pts with t(8;21)
Micro: immature eosinophils in the BM show large purple-violet granules that may obscure cell morphology; these abnormal eosinophils may show abnormal nuclear hyposegmentation
- the abnormal granules may help identify inv(16) although it is not specific and has been seen in AML with biallelic CEBPA mutations and early T-cell precursor ALLs [7]
IHC: blasts CD34/CD117+, with differentiation towards granulocytic lineage, CD13/33/15/65/MPO+
- often CD2+, but this is not specific
Genes: inv(16)p13.1q22) is the most common, t(16;16)(p13.1;q22) is less common though both cause fusion of CBFB at 16q22 to MYH11 at 16p13.1
- MYH11 codes for smooth muscle myosin heavy chain
- CBFB codes for beta subunit of core binding factor
- may be subtle and missed by KT, so FISH or PCR may be needed
- secondary trisomy 22 is fairly specific for inv(16)
Px: high rate of complete remission and favorable overall survival
Blasts in inv(16) or t(16;16) - with abnormal cytoplasmic inclusions [4]

AML with inv(16), blasts were tryptase+ [7]

.jpg?crc=271853780)
Acute myeloid leukaemia with inv(16)
(p13.1q22). Abnormal eosinophils, one with large basophilic coloured granules, are present.
Acute Promyelocytic Leukemia (APML or APL) with t(15;17)(q22;q12) PML-RARA
- FAB M3
- a Diagnostic Emergency!
> 20% (blasts + abnormal promyelocytes)
• Hypergranular and microgranular types
• DIC, life threatening coagulopathy
High value of cytochemical MPO and morphology in diagnosis
- if WBC low: search for hypergranular promyelocytes
- if WBC high: consider microgranular APL every time there are folded nuclei, monocytic
Micro: rare cases do not resemble either hypergranular or microgranular APL
IHC: Myeloid antigens (+); CD34(-), HLA-DR(-) CD15(-)
– Do not diagnose by flow
Genes: minority of cases cryptic (negative) by cytogenetics and FISH but are PCR positive
Tx: All trans retinoic acid (ATRA) + conventional chemo
- Early ATRA essential to reduce risk of hemorrhage
- takes about 5 days of ATRA to decrease risk
Acute Promyelocytic Leukemia (APL)

Flow cytometry of Acute Promyelocytic leukemia. Blasts have high side scatter and can be confused with maturing granulocytes. MPO+, CD34 negative, and HLA-DR negative, CD117+, CD13/33+ [4]

Translocations considered diagnostic in APML [Atch Pathol Lab Med. 2015 Oct;138(10):1308-13]
AML with t(9;11)(p21.3;q23.3); KMT2A-MLLT3
• 11q23 KMT2A gene used to be MLL gene
• Monocytic differentiation, Intermediate risk;
- More common in kids
– 9-12% pediatric AML
– 2% adult AML
• Secondary genetic abnormalities are common: +8
most common
AML with t(6;9)(p23;q34.1); DEK-NUP214
In up to 2% of AML cases, in both kiddos and adults, median age 13 and 35-44 yo
- presents c anemia and TBCpenia, generally lower WBC count in adults
- Auer rods present in 1/3 of cases
• PB basophilia is a characteristic feature
• Multilineage dysplasia, esp erythroid and granulocytic
IHC: (+) MPO, CD9, CD13, CD33, CD38, CD123, HLA-DR, CD117 (usually), CD34 (most), CD15, CD64 (some)
- 50% are TdT+
Genes: DEK on cr 6, NUP214 (aka CAN) on cd 9, fusion alters nuclear transport by binding transcription factors
Px: poor, esp if presenting with higher WBC counts
- high freq FLT3-ITD, tho does not negatively affect px in kiddos
AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM
• MECOM used to be EVI1
- Should consider history of MDS, if has MDS-related cytogenetic abnormality, morphologic evidence of dysplasia in >50% of cells in 2 lineages, as well as the absence of prior cytotoxic or radiation therapy, and if has a recurring cytogenetic abnormality
Micro:
Dysplastic small hypolobated megakaryocytes
• Multilineage dysplasia
Ddx: CML blast phase, AML with MDS-related changes (AML-MRC)
Px:
• Poor risk type of AML
AML with (megakaryoblastic) with t(1;22)(p13.3;q13.1); RBM15-MKL1
• Restricted to infants and children <3 years old without Down syndrome
• Megakaryob lasts
Micro: PB and BM similar to that of acute megakaryoblastic leukemia (a subtype of AML, NOS) megakaryoblasts may show cytoplasmic blebbing or pseudopod formation
IHC: express one or more of these: CD41, CD61, CD42b
- CD36 is characteristically positive, but may be negative
- CD13 and CD33 may be positive
- CD34, CD45, and HLA-DR often are negative
- MPO is negative
- TdT and lymphoid markers are not expressed
Genes: t(1;22)([13.3;q13.1) is the sole abnormality
DDx:
– Myeloid leukemia associated with Down syndrome
– Another type of megakaryoblastic leukemia (CD41/CD61+) seen in infants and children
– Does not show t(1;22)
– GATA1 mutations
– Transient abnormal myelopoiesis
– >10% blasts, age 3-7 days, which resolve without treatment, GATA1 mutations
Acute Megakaryoblastic Leukemia
AML with BCR-ABL1
- Provisional entity, rare (<1% of AMLs)
- vs CML, less often splenomegaly, lower PB basophilia, fewer dwarf megs
This disease is a De novo AML in which patients show no evidence (either before or after therapy) of chronic myeloid leukemia
- Cases of mixed phenotype acute leukemia (MPAL) and therapy-related are excluded
- If another recurring genetic abnormality is present, that abnormality drives the final classification
Deletion of antigen receptors (IGH, TCR), IKZF1 , and / or CDKN21 may support a diagnosis of de novo disease
Tx: Patients may benefit from tyrosine kinase inhibitor therapy
AML with BCR-ABL1. Must exclude CML
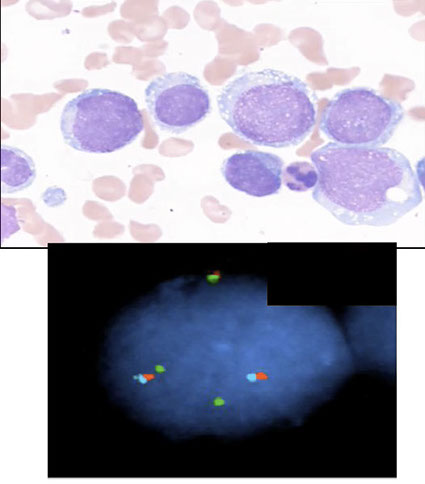
AML with gene mutations
-
AML with mutated NPM1
– Favorable prognosis only in absence of FLT3 ITD mutation; 30% of AML
- secondary mutations include FLT3 and DNMT3A
Micro: strong association with acute myelomonocytic leukemia and acute monocytic leukemia (80-90% of acute monocytic leukemias have NPM1 mutation
- Multilineage dysplasia found in 74/318 (23%) de novo NPM1 mutated AML
- No significant survival difference of multilineage dysplasia in the presence of NPM1 mutation, a normal karyotype and no history of MDS
- Presence of NPM1 mutation trumps multilineage dysplasia
- secondary AML cases (arising from MDS, MPN, therapy related) lack favorable prognosis of de novo AML
__________________________________________
IHC: high CD33, variable (often low) CD13, usually positive for CD117, CD123, CD110
- negative HLA-DR and CD34 (CD34+ cases associated with a worse prognosis)
2 flavors described
1. Immature myeloid IHC profile
2. Monocytic profile (CD36+, CD64+, CD14+)
CD34+/CD25+/CD123+/CD99+ associated with FLT3 mutations
__________________________________________
Genes: Usually normal karyotype
- - up to 15% have abnormal KT, such as gain 8 and del(9q) --- del(9q) associated with AML-MRC
- If there are other MDS-related cytogenetic changes, should diagnose MDS-MRC even if NPM1 is mutated
Cases with RUNX1 and NPM1 mutations should be classified as AML with mutated NPM1
- NPM1 commonly seen as a secondary change in AML, esp with FLT3 and DNMT3A, TET2, IDH1/2
- NPM1 mutations appear to preceed FLT3 mutations
__________________________________________
Px: good response to induction therapy
- cases with normal KT, and absence of FLT3-ITD have a favorable px
- worse px if FLT3-ITD present in younger pts, not clear if FLT3-ITD mutations give poorer px in older pts
- co-ocurrence of FLT3, NPM1, and DNMT3A is particularly poor

AML with NPM1 mutations, nuclei have cup-like indentations

AML with NPM1 with high Variant Allele Frequency (VAF) do worse than those with low VAF [Blood. 2018 Jun 21;131(25):2816=25]

AML with mutated CEBPA
7-20% of AMLs have CEPBA mutations
- more frequent with normal karyotype
-12-47% are single/monoallelic
– Favorable prognosis, only in 10% of AML, although ~50% of adult cases with normal karyotype
- strong assoc c monocytic differentiation
– Cytogenetically normal AML and AML with 9q deletion
- Multilineage dysplasia found in 28/108 (26%) of CEBPA mutated AML patients
- No significant survival difference with multilineage dysplasia
Survival in AML with CEBPA, Wouters BJ, et al. Blood. 2009 Mar 26;113 13):3088-91.
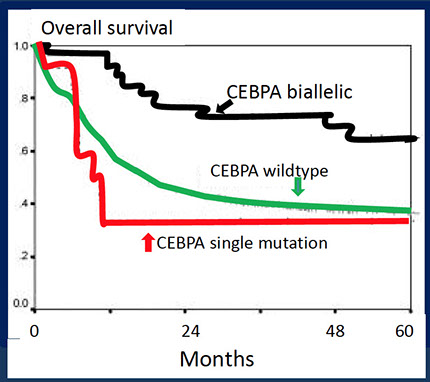
AML with biallelic mutations of CEBPA
Biallelic mutations required for this category
- 7-20% of all AML cases have a CEBPA mutation
Approximately half are biallelic
Immunophenotyping: increased frequency of CD7 and CD15 expression on the blasts
- monocytic markers (CD14, CD64) usually absent
- important to recognize if it is a germline mutation, bc may be important in transplantation
Genes: low freq of karyotype abnormalities
- del(9) sometimes seen, doesn't change px
NGS reporting clues:
Classic pattern, frameshift or non-sense variant in the N-terminus (p.1-120) and an in-frame indel or missense variant in the C-terminus (p.278-358).
- Should raise the possibility of germline predisposition condition
Px: favorable

No unique morphologic features; 25% cases may show multilineage dysplasia AML with biallelic mutations of CEBPA
AML with mutated RUNX1
- Gene located at 21q22
- encodes the alpha subunit of the core binding factor
- Present in 12-13% of AML
- more freq in older male pts
- frequent prior hx of MDS, or prior exposure to radiation
*** Cases arising from MDS will still be called AML-MRC [4] ***
*** Cases with prior therapy will still be therapy-related AML ***
Micro: wide morphologic spectrum
Genes: freqently assoc c KMT2A-PTD, IDH1, IDH2, or ASXL1 mutations
- rare CEBPA or NPM1 mutations
Px: poor response to therapy with shortened survival
- germline mutations should be evaluated, think about it in younger pts

Wide morphologic spectrum;
No unique morphologic features
AML with myelodysplastic changes
Diagnostic criteria:
20% BM or PB blasts + (one of the following):
-Hx of MDS
-MDS cytogenetics
- Multilineage dysplasia (>50% in 2+ cell lineages, which is HIGHER than in regular MDS)
all these WITHOUT:
-prior cytotoxic therapy for any dz
- absensce of the specific cytogenetics abnormalities seen in AML with recurrent genetic abnormalities
MC in elderly
Usually have severe pancytopenia
May appear de novo or from an evolving MDS
________________________________
IHC: w chr 5/7 del get (+) CD34, 7, TdT
________________________________
Genes: del -5 and del -7 are MC
- If NPM1 mutation identified, diagnose as AML with NPM1 mutation (despite MLD)
- If biallelic CEBPA mutations, diagnose as AML with CEBPA mutation (despite MLD)
- If AML-MRC diagnosis is based on history of MDS or MDS-related cytogenetics, then retain AML-MRC even if NPM1, biallelic CEBPA mutations identified
Del(q) assoc c t(8;21), freuently occurs in AML with NPM1 and biallelic CEBPA mutations
SF3B1 mutations are not associated with ring sideroblasts in AML as they are in MDS
- although ring sideroblasts in AML are associated with adverse risk characteristics and have a distinct gene expression pattern
________________________________
DDx: refractory anemia w excess blasts, acute erythroid leukemia, acute megakaryoblastic leukemia, other AML NOS
________________________________
Px: Poorer than most AML

AML genes [4]
Morphologic dysplasia in AML [4]

Red box is genes assoc c MDS or CMML, includes SRSF2, ZRSR2, SF3B1, ASXL1, BCOR, EZH2, U2AF1, STAG2, R Coleman Lindsay et al. Blood 2015; 125:1367-76)


Micromegakaryocytes and Hypogranulated myeloid cells independently associated with shorter OS/EFS in multivariable analysis [4] [Weinberg et al, Mod Pathology 2015 28(7):965-76
Therapy-related myeloid neoplasms
Pts who develop myeloid neoplasms following cytotix therapy
- 70% treated for slid malignancy and 30% for hematologic malignancy
- common occurs 5-10 years after exposure to alkylating agents and/or ionizing radiation
- 1-5 years following tx with agents that interact with DNA topoisomerase II (topoisomerase II inhibitors)
- AML and MDS grouped togerher
- may have recurring cyogenetic abnormalities that impact px and hsould be noted in the dx
- may occur after therapy for another AML type
Includes:
1. therapy-related acute myeloid leukemia (t-AML)
2. t-MDS,
3. therapy-related MyeloDysplastic / MyeloProliferative Neoplasms (t-MDS/MPN)
- treated (?) MPN transformation not included bc cant know if its from tx or primary (?)
- classification based on morphology and blast counts may not have a clinical significance
All agents will usually cause genetic abnormalities
2 subtypes:
1. Alkylating agents (cyclophosphamide, busulfan)
- causes MDS, then BM failure, then AML ~5 years after tx
-- most pts die in MDS phase
2. Topoisomerase II inhibitors (etoposide, doxorubicin)
- see monocyte proliferation about 2 years after tx
IHC: no specific immunophenotype exists
Risk of acquiring one of these dzs inc w age w tx, w exception of etoposide
- may be caused from tx of any kind of malignancy, usually 5-10 years after drug exposure
Dysplasia occurs in multiple lineages
Unbalanced chromosome 5/7 usually the culprit, but translocations may be balanced
Anemia and unusual RBCs usually present as well, + ringed sideroblasts, dyserythro-/-granulopoiesis, neutos / basos / megakaryos usually strange looking
Genes: 90% have abnormal karyotype
- 70% unbalanced, usually partial loss of chr 7 or 5
- remaining % have balanced translocations
Prog: Generally poor (<10% 5-year); very poor chr 5/7 probs
- depends a lot on primary dx and tx received

AML, NOS
AML's that don't fit the other categories, still have the >20% blasts req'd for AML, can be further classified based on features
- may need further genetic / molecular testing to be categorized; these may be subcategorized based on IHC / morphology (degree of differentiation or cell type), but genetics is best px indicator
AML with minimal differentiation
Rare; this disease is an AML with no morphologic or cytochemical evidence of myeloid differentiation
- the myeloid nature of the blasts is seen with immunological markers, which is essential when distinguishing from a lymphoblastic leukemia
- does not fulfill criteria for other types of AML
- usually in infants or older adults
Micro: blasts are small and undifferentiated
IHC: most cases express CD34, CD38, and HLA-DR - blasts express at least 2 myeloid markers including CD13 and CD117
- negative for myeloid and monocytic markers such as CD11b, CD15, CD14, CD65
- negative for MPO by cytochemistry, but can be positive for MPO by IHC or by flow
- nuclear TdT positive in 50% of cases, and thought to be a favorable prognostic indicator
Px: probably the worst of this disease category
AML without maturation
5-10% of AML cases; High percentage of BM blasts without significant evidence of maturation to more mature neutrophils
- maturing cells of the granulocytic lineage constitute <10% of nucleated BM cells
- myeloid nature of cells confirmed by MPO staining or presence of Auer rods
IHC: positive for MPO and one or more myeloid antigens, such as CD13, CD33, or CD117
- CD34 and HLA-DR are positive in ~70% of cases
- generally there is no expression of markers associated with granulocytic maturation (CD15 or CD65) or monocytic differentiation (CD14, CD64)
- CD11b expressed in some cases
- CD7+ in 30% of caes
- lymphoid markers (CD2, CD4, CD19, CD56) seen in 10-20% of cases
DDx: lymphoblastic leukemia in cases where blasts lack granules of have low percentage of MPO-positive blasts
AML with maturation
~10% of AML cases; characterized by 20% blasts in BM or PM and with evidence of maturation (>10% maturing cells of granulocytic lineage) in BM; cells of monocyte lineage are <20% of BM cells
- does not fit into another category of AML
- pts usually present with anemia, thrombocytopenia, and neutropenia
- WBC and # blasts is variable
Micro: Auer rods frequently present
- promyelocytes, myelocytes and mature neutrophils are >10% of BM cells
- dysplasia is frequently present, although <50% of cells in two lineages are dysplastic
- eosinophil precursors are frequently increased, but are not abnormal (as seen in the inv(16) or t(16;16))
IHC: blasts usually express one or more myeloid markers, such as CD13, CD33, CD65, CD11b, CD15
- there is usually expression of CD34, CD117, and HLA-DR, may only be present in some blasts
- monocytic markers (CD14, CD36, CD64) usually are negative
- CD7+ in 20-30% of cases
- expression of CD56, CD2, CD19, and CD4 is uncommon
DDx: MDS-EB, AML without maturation, AMML
Acute Myelomonocytic Leukemia (AMML)
aka AML M4, 5-10% of AML cases, more common in older people
• Both myeloid & monocyte precursors
– >20% blasts: Blast count includes promonocytes
– >20% neutrophils and myeloid precursors
– >20% monocytes and promonocytes
Micro: monoblasts are large cells with lots of cytoplasm that can be basophilic and can show pseudopod formation
- azurophilic granules, vacuoles and Auer rods may be seen
IHC: generally shows several populations of blasts, which variably express myeloid antigens (CD13, CD33, CD65, CD15), with one of the blast populations positive for monocytic markers (CD14, CD64, CD11b, CD4, CD36, CD68, CD163, lysozyme)
- co-expression of CD15, CD36, and strong CD64 is characteristic of monocyte differentiation
- there is also a population of immature blasts expressing CD34 and or CD117
- most cases positive for HLA-DR and ~30% of cases express CD7
Genes: Non-specific
DDx: AML with maturation, acute monocytic leukemia, chronic myelomonocytic leukemia
Acute Monoblastic and Monocytic Leukemia
Acute monoblastic leukemia has >80% monoblasts
- in acute monocytic leukemia, most of the leukemic cells are promonocytes or monocytes
Extramedullary masses, cutaneous and gingival involvement, and CNS involvement are common
Micro: Auer rods are rare, when present
- hemophagocytosis may be seen, and is often assoc with t(8;16)(p11.2;p13.3)
IHC / flow: (+) CD13, CD33, CD15, CD65
- generally has at least two markers of monocytic differentiation: CD14, CD4, CD11b, CD11c, CD64 (bright), CD68, CD36 (bright), and lysozyme
- CD34 is positive in only 30% of cases, CD117 is more often expressed
- most cases are positive for HLA-DR
- MPO expressed in acute monocytic leukemia, less often in monoblastic leukemia
- aberrant CD7 and / or CD56 in 25-40%
Genes: t(8;16)(p11.2;p13.3) in most cases is assoc c hemophagocytosis
DDx: AML without maturation, AML with minimal differentiation, AML with t(9;11)((p21.3;q23.3), acute megakaryoblastic leukemia, extramedullary myeloid sarcoma
- DDx of acute monocytic leukemia includes CMML, AMML, microgranular APL
Acute monoblastic and monocytic leukemia [4]

Pure erythroid leukemia
- (Acute Erythroid Leukemia removed from WHO and now MDS with excess blasts and an erythroid preponderance??)
Denominator for blast calculation is now all nucleated cells
- need >80% of all bone marrow cells to be erythroid precursors, with >30% proerythroblasts
- used to be non-erythroid cells when erythroid cells >50%
- erythroid/myeloid acute erythroid leukemia -> MDS with excess blasts
- cases with >50% erythroid cells and >20% myeloid blasts should be classified as AML-MRC (Myelodysplastia-Related Chenges) or AML-RGA (Recurrent Genetic Abnormalities)
- AML M6;
• Proposed WHO Revision to move this to a myelodysplastic syndrome, since it has a more mild clinical course
Micro: Erythroid hyperplasia and dysplastic erythroid
precursors; PAS positive; cytoplasmic vacuoles
• Erythroleukemia M6a
– > 50% of all nucleated cells erythroid cells
– >20% of non-erythroid cells are myeloblasts
• Pure erythroid leukemia M6b (rare): >80% erythroblasts
Cytochemical: NSE and MPO negative
IHC: (+) E-cadherin (more specific for erythroids), CD117, erythroiblasts express glycophorin, hemoglobin A, and CD36 and CD71 (less specific)
- glycophorin and hemoglobin may be negative if less differentiated
Negative immunostains: CD3, CD20, CD138, PAX5, TdT, CD34, CD10, HLA-DR, Myeloperoxidase, Lysozyme, CD14, CD61, Cytokeratins (OSCAR, AE1/3), CD56, Desmin
Px: Poor, 3 mo survival
Pure erythroid leukemia
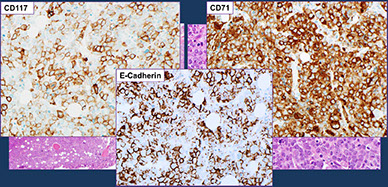
Blast counting in erythroid predominant cases. Reichard lecture, city of hope, LASOP, 1/18/2020

Pure erythroid leukemia. [5]

Pure erythroid leukemia, IHC is e-cadherin

Erythroid immunophenotype

Acute Megakaryoblastic Leukemia
- AML M7
• > 20% blasts, 50% are megakaryoblasts
• Excludes t(1;22), inv(3), t(3;3) and Down syndrome-related cases
- de novo AML restricted to infants and young children (aged <3 years), with most cases occurring in the first 6 months of life
Micro: BM fibrosis; Basophilic cytoplasm; Cytoplasmic blebs
- small and large megakaryoblasts may be present and mixed with more normal blasts
EM—platelet peroxidase
– CD41, CD61, CD42
acute megakaryoblastic leukemia, right shows extensive fibrosis [4]

acute megakaryoblastic leukemia aspirate with prominent pseudopod formation[4]

acute megakaryoblastic leukemia, CD117 (left) and CD61 (right), higlighting blasts

Acute basophilic leukemia
Very rare, blasts differentiate towards basophils
- patients may have cutaneous involvement, organomegaly, lytic lesions, and sx related to hyperhistaminemia
Micro: blasts in PB and BM are medium-sized, high NC ratios, 1-3 prominent nucleoli, moderately basophilic cytoplasm with variable numbers of coarse basophilic granules, may have vacuoles in cytoplasm
- erythroid series can show some dysplasia
- metachromatic staining with toluidine blue
IHC: blasts express CD13 and CD33 and are usually positive for CD123, CD203c, and CD11b
- negative for monocytic markers and CD117;
- may be positive for CD34
- unlike normal basophils, may be positive for HLA-DR but are negative for KIT
DDx: blast phase of an MPN; other AML subtypes with basophilia such as AML with t(6;9); AML with BCR-ABL1; mast cell leukemia; and rarely subtypes of lymphoblastic leukemia with prominent coarse granules
- mast cell leukemia will be positive for CD117, MCT, and CD25
Acute panmyelosis with myelofibrosis (APMF)
A very rare form of AML in which there is increased erythroid, granulocyte, and megakaryocyte precursors (ie panmyelosis)
- patients are acutely sick at presentation, with weakness, fatigues, and usually fever and bone pain
- Pancytopenia is ALWAYS present
- no or minimal splenomegaly
- clinical evolution is rapidly progressive
Micro: pancytopenia in the PB, which is usually marked, possible rare erythroblasts, no dacryocytes, may see occasional neutrophil precursors or blasts
- although dysplastic changes in myeloid cells is frequent, criteria for AML with myelodysplasia-related changes is not met
BM aspiration is usually not successful
- BM core biopsy is hypercellular, with densely fibrotic stroma, and panmyelosis
- a characteristic finding is foci of blasts associated with dysplastic megs that are small with eosinophilic cytoplasm
- blasts usually range from 20-25%
- degree of marrow fibrosis is variable, but is usually marked, and diffuse collagenous fibrosis is less common
IHC: blasts are CD34+, CD13/33/117+
- MPO usually negative
Genes: usually abnormal cytogenetics,
- if complex karyotype with cr5 or 7, consider the dx of AML-MRC
DDx: other types of AML associated with BM fibrosis, including acute megakaryoblastic leukemia
- PMF, post-polycythemia vera myelofibrosis, post-essential thrombocytemia myelofibrosis
- the differential may be difficult especially if no cytogenetics can be obtained
- if the proliferative process is predominantly one cell type (ie myeloblasts) and there is associated myelofibrosis, then should be classified as AML with a specific subtype and qualified as "With myelofibrosis"
- blasts of acute megakaryoblastic leukemia are CD34 negative
Px: Poor, only survival of a few months
Myeloid sarcoma
Older adults (mean age 56 years, range 1 month to 89 years); M:F 1.2:1., 10% of pediatric AML.
•Sites of involvement: skin, LN, GI, bone, ST, testes, CNS, orbit, etc., Multiple sites in <10% cases.
•Detection of myeloid sarcoma is equivalent to a diagnosis of AML (new or relapse).
•May occur as therapy-related myeloid neoplasm
Tumor with myeloblasts outside the BM, but must form a mass (can't just be infiltrating metastatic cells) in any, or multiple sites
- should be subtyped
Micro: Blasts may show myeloblastic (+/- admixed maturing granulocytes), promyelocytic, myelomonocytic, or monoblastic differentiation.
•Rarely erythroblastic or megakaryoblastic morphology (more common in blastic transformation of MPNs).
•LN shows paracortical or diffuse involvement, often extending to the perinodal fat.
•May mimic metastatic carcinoma or lymphoma.
May precede or coincide with AML, MDS, or cancer recurrence
55% have genetic abnormalities
Chromosomal aberration in about 55% of the cases (monosomy 7, trisomy 8, KMT2A/MLL rearrangement, inv(16), trisomy 4, monosomy 16, loss of 16q, 5q, or 20q, trisomy 11).
•NPM1 (16%) and FLT3 (<15%) mutations.
•All MS cases should be classified immunophenotypically and cytogenetically (karyotype and/or FISH)
–may be done on BM/PB if involved.
–on extramedullary tissue otherwise.
11q23 (KMT2A/MLL) rearrangement
•Myelomonocytic morphology.
•39% of MS cases in children.
•Higher incidence of skin involvement, esp. pediatric.
t(8;21)(q22;q22) RUNX1/RUNX1T1
•25% of pediatric and 10% adult cases develop MS (head and neck localization in pediatric pts but not adult cases).
•CD19/PAX5 aberrant expression common.
Trisomy 8
•Higher incidence of skin or breast involvement.
NPM1 mutation
•CD34 negative, aberrant cytoplasmic NPM1 expression.
•Myelomonocytic or monoblastic morphology,
•Normal karyotype.
•Higher incidence in MS involving the skin (>50%).
CBFB-MYH11 Fusion (inv16 or t(16;16) prevails in the abdomen
- Myeloblastic or myelomonocytic differentiation.
•Many cases (64%) lack overt eosinophilic component.
•Striking abdominal (GI, GU) localization [100% (11/11) cases reviewed at Mayo Clinic Rochester; overall 94% (31/33) in published cases).
•Breast involvement also common.
•Plasmacytoid dendric cell (pDC) nodules in a subset of cases (CD123+ but CD56-).
DDx: Lymphoma (LBL, blastoid MCL, BL, DLBCL).
•Blastic plasmacytoid dendritic cell neoplasm.
•Non-effacing extramedullary blastic proliferations.
•Extramedullary hematopoiesis (advanced stage MPN).
•Pseudotumoral proliferations following growth factor administration.
•Small round cell tumors; carcinoma.
MPO and CAE are (+), whereas NSE is (-) (differentiating from monoblasts [which are NSE(+), and also CD14/163/11c(+)])
CD68/KP1 is MC IHC expressed, CD117/99 also common
- may see CD123(+), indicating plasmacytoid dendritic cell differentiation
- aberrant markers also seen
Tx: BMT assoc c highest survival/cure rates
Myeloid sarcoma





from lecture at City of Hope, January 18, 2020


Myeloid sarcoma with CBFB-MYH11

Myeloid proliferations related to Down syndrome
50x increased risk of leukemia from Down Syndrome in first 5 years (affecting 1-2% of DS pts)
Blast count foes not drive the prognosis, and thus should not be classified as AML or MDS
- majority (50%) are acute megakaryoblastic leukemia, GATA1 (+) [vs other AMkbLs], and (+) for CD34, CD7, CD33, CD41, CD61, CD71, CD117, and glycophorin A, variably (+) for CD4 (dim), CD13, CD56
~ 10% of neonates with Down Syndrome have some type of hematologic abnormality at birth (transient myeloproliferative disorder) aka transient abnormal myelopoiesis (TAM), which is indistinguishable from AML and resolves within several months but may predispose to leukemia later on
- MC thrombocytopenia in blood work
Cells often have dark basophilic cytoplasm with granules and cytoplasmic blebbing (suggestive of Mkc's)
GATA1 (+) in blasts, and is most likely different from AML blast genetics
Refractory Cytopenia of Childhood often preceeds MDS or leukemia
- Macrocytic erythrocytes and giant platelets can be seen
IHC: CD34/56/41 (+) in Transient Abnormal Myelopoiesis, but negative in 50% of leukemias
AML in DS pts assoc c better px than children w/o DS that have AML
- specific protocols exist for these GATA1 (+) leukemias
Px: Myeloproliferative disorder that spontaneously resolves within 1-2 months
- Transient Abnormal Myelopoiesis
Myeloid proliferations assoc c Down syndrome [4]


Blastic plasmacytoid dendritic cell neoplasm (BPDCN)
BPDC comes from plasmacytoid dendritic cell precursors (pDCs or plasmacytoid monocytes) which form blast-filled tumors that like to spread to lymph nodes
- rare tumor mostly in elderly, assoc c MDS
Almost always involves skin, ~80% in PB/BM, ~45% in LNs
Presents c single/multiple skin lesions, sometimes lymphadenopathy (20%), thrombocytopenia, and usually will develop into leukemia
Micro:
Lymph node
•Interfollicular involvement sparing the follicles.
Bone marrow
•Mild interstitial infiltrate or massive involvement.
•Blastic cells may show microvacuoles and pseudopods with agranular cytoplasm.
PB/BM smear shows pseudopods and membranous microvacuoles
Must distinguish from myeloid neoplasm extensively involving a particular site
IHC: (+) CD4/7/33/43/45RA/56/123/303, TCL-1A, CLA, MXA (IFN-α inducible protein myxovirus A), Tdt
- cannot r/o if 56 negative and 4/123,TCL-1 (+)
- CD68 in 50% (will see small cytoplasmic dots)
- Granzyme B, perforin, and TIA1 found in mRNA but negative on immunostains
- must do extensive IHC/genetic analysis before Dx'ing
May express myeloid (CD33, CD36, CD68) or lymphoid (CD2, CD5, CD7, CD38, CD79a) markers, CD117, S100 (30%), TdT (30%, variable expression).
•Negative for CD3, CD19, CD20, lysozyme, MPO.
•Combination of CD4, CD56, CD123, and TCL1A; in the absence of lineage specific markers highly specific for BPDCN.
MPO and NSE are negative
DDx: Acute myeloid leukemia/myeloid sarcoma.
•Mature plasmacytoid dendritic cell proliferation/MPDCP (pDC nodules).
•Lymphoblastic leukemia/lymphoma.
•T/NK-cell lymphoma.
•Langerhans cell histiocytosis.
DDx: Myeloid sarcoma - See the table
Frequently show skin and/or involvement, esp. cases with monocytic differentiation.
•May express CD4, CD56, and/or CD123.
•Shares expression of many other myeloid antigens with BPDCN (CD14, CD33, CD43, CD68, CD163).
•Extensive IHC workup and genetic analysis may be required for distinction between BPDCN and MS.
DDx: MPDCP/pDC nodules - See image and table
Nodules or irregular aggregates of morphologically mature cells morphologically and phenotypically similar to normal pDCs (CD56 negative or focal/weak, TdT negative).
•May be seen in LN, skin, and BM.
•May be numerous/confluent or “tumor forming.”
•Often associated with and clonally related to the underlying myeloid neoplasms (chronic myelomonocytic leukemia/CMML, MDS, AML).
•Also seen in reactive conditions: Kikuchi-Fujimoto lymphadenitis, SLE, hyaline-vascular Castleman disease, granulomatous lymphadenitis.
Genes:
Two thirds of cases show abnormal karyotype (nonspecific; 5q21/5q34, 12p13, 13q13-21, 6q23-qter, 15q, -9/CDKN2A deletion).
•Complex karyotype common.
•TET2: most commonly mutated gene in BPDCN.
•Other mutations detected: ASXL1, RAS, ATM, IDH2, KIT, APC, RB1, BRAF, TP53, RET.
Complex karyotype mixing is common
Tx: Acute leukemia-type induction with possible BMT
Initial response to chemotherapy followed by relapses (skin, other sites) and chemoresistance.
•10-20% associated with or develop into other myeloid neoplasms (CMML, MDS, AML).
•Aggressive; median survival 10-20 months.
•Better in pediatrics pts; long term survival in 36%.
Px: Aggressive with ~1 year avg survival
- tend to relapse after initial response to chemo
BPDCN in skin, diffusely infiltrating dermis with perivascular pattern of infiltration with fine chromatin resembling lymphoblasts or myeloblasts

Characteristic phenotype is CD4+, CD56+, CD123+

BPDCN IHC summary table





MYELOID SARCOMA

Acute Leukemias of Ambiguous Lineage
Overview of Acute Leukemias of Ambiguous Lineage (ALAL)
Leukemias that don't show maturation along a single lineage, shown by FC
- may have no lineage-specific antigens (Acute Undifferentiated Leukemia, AUL), or express antigens from more than 1 lineage (Mixed Phenotype Acute Leukemia, MPAL)
For MPALs, a bilineage leukemia has separate blast populations from more than 1 lineage, and biphenotypic leukemia is one blast population that contains antigens from multiple lineages
- some examples of these biphenotypic leukemias are B/myeloid (B/MY) and T/myeloid (T/MY)
-- FGFR1 mutations are not to be assoc c T/MY
Criteria for recognizing specific cell lineages in ALAL
3 ways in recognizing cells from myeloid lineage:
1.) 2 distinct blast populations (1 of which would qualify as AML [though doesn't need to have 20% blasts {?}])
2.) Single blast population that would qualify as B-ALL or T-ALL (c (+) lymphoid markers), and are MPO (+)
3.) " " and show monocytic markers (CD11c/14/36/64)
Cells from the T-lineage are recognized c surface or cytoplasmic CD3 (+)
- must be careful bc NK-cells can stain positive c CD3 due to the zeta-chain of their T-cell receptor
B-cell lineage identified with either strong- or weak- CD19 positivity and 1- or 2- other B-cell antigen (respectively) strong positivity (CD10, CD22 [cytoplasmic], or CD79a)
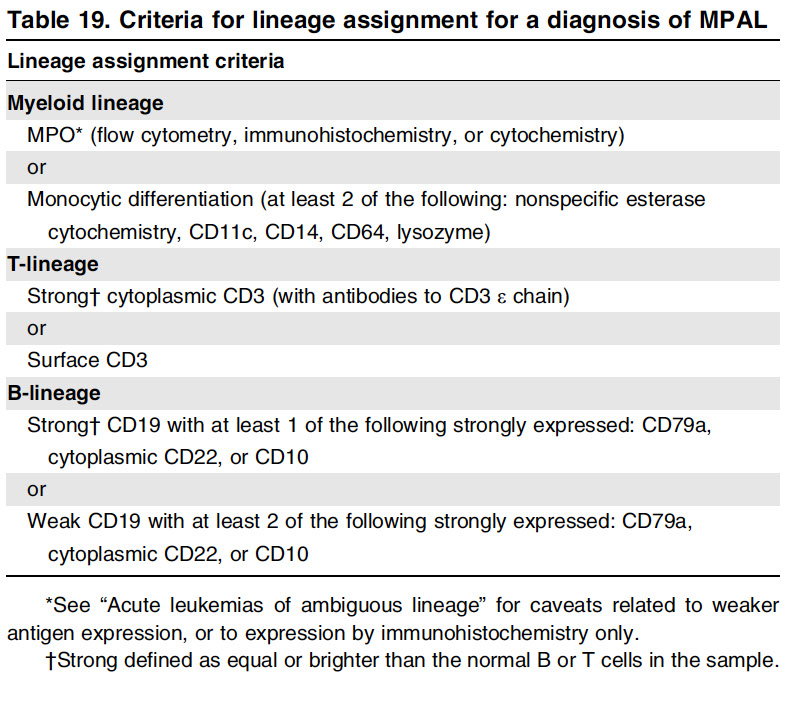
Acute undifferentiated leukemia
Includes leukemias that express no markers for either the lymphoid or myeloid lineages
- must exclude rare lineages (ie plasmacytoid dendritic cell / NK-cell / basophilic / non-hematopoietic cell) before diagnosing
Blasts will be negative for MPO and esterase, CD3/19/22/79a
- blasts can have (+) CD34/38 or TdT
Cases are too few to know exact Px
Mixed phenotype acute leukemia with t(9;22)(q34;q11.2): BCR-ABL1
Meets the criteria for MPAL (states in the ALAL overview) and has t(9;22)(q34;q11.2) or BCR-ABL1 rearrangements
Dx should not be made in pts c known CML
MCC of MPAL, though still accounts for <1% of acute leukemias
All cases have at least the t(9;22)(q34;q11.2) or BCR-ABL1 abnormalities, though complex karyotypes also occur
Majority of these cases have blasts from B-cell and myeloid lineage; less come from T-cell / myeloid lineages; and even fewer from all 3 lineages
Imatinib and tyrosine kinase inhibitors would be expected to treat these kinds of pts, though no good data exists
Px is poorer than other types of MPALs
Mixed phenotype acute leukemia with t(v:11q23): MLL rearranged
Meets the MPAL criteria and has the above translocation
As with AML/ALL c MLL rearrangements, this leukemia is MC in infancy
Usually have a population of monoblasts c a population of lymphoblasts
If the blasts are only monoblasts (w/o lymphoblasts), consider AML c MLL translocation
Lymphoblasts are usually CD15/19 (+) and CD10 (-)
- CD22/79a is usually weak
In addition to MLL rearrangement, t(9;11) and t(11;19) have been seen
Poor Px
Tx is uncertain
Mixed phenotype acute leukemia, B/myeloid, NOS
Blasts express the B and myeloid markers, as in the criteria set in the overview, but do not have any specific / characteristic cytogenetic abnormalities
This group has no characteristic cytogenetic abnormalities, and the blasts display no unique features morphologically
- may have relatively simple to complex karyotypes
Px is poor
Genetic abnormalities found are usually unfavorable, which may explain the poor outcome
Mixed phenotype acute leukemia, T/myeloid, NOS
Meets criteria for T-cell and myeloid lineages, as described in the overview, but has no characteristic cytogenetic abnormality
Blasts have no distinct morphologic features
Myelo-/monoblasts are CD3/13/33/117 (+) and T-cells CD2/3/5/7 (+)
Early T-precursor (ETP) ALL leukemia has early T cell diff with retention of some myeloid and stem cell characteristics at immunophenotypic and genetic level
- must express CD7 but lack CD1a and CD8, and are positive for 1 or more of the myeloid / stem cell markers CD34, CD117, HLA-DR, CD13, CD33, CD11b, or CD65
- express CD2 and cytoplasmic CD3 and often CD4
- CD5 often negative
- myeloid gene mutations such as FLT3, NRAS/KRAS, DNMT3A, IDH1, and IDH2 found at high freq whereas T-cell assoc mutations such as NOTCH1 or CDKN1/2 mutations infreq
No characteristic genetic defects due to low number of cases
Poor px
No tx regime has been established
Mixed phenotype acute leukaemia, NOS- rare types
Few cases of trilineage leukemias (B/T/myeloid) have been reported
No cases of (B/T)/Mgkc or (B/T)/erythroid leukemias have been reported, maybe bc Mgkc's and erythroid lineages are the first to break off the pluripotent stem cells
Assessing MRD in AML?
[Blood 2018 Mar22;131(12):1275-91]
Although considerable variations in techniques and testing methodologies exist, detection of MRS is a valuable tool for prediction of relapse in AML
Methods include:
- real-time PCR
- flow cytometry
- next generation sequencing (NGS)
Outside of a clinical trial, routine adoption of MRD assessment in the upfront management of AML is not yet recommended
- efforts to standardize MRS are underway
Reasons for inadequacies in measuring MRD

MRD methods [4]


References
1. Arber et al. The 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia. Blood. 19 May 2016. Vol 127, No 20.
http://www.bloodjournal.org/content/bloodjournal/127/20/2391.full.pdf
2. Hasserjian R. MDS and MPN, Part 1: Into to Myeloid Neoplasms. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=416053016
3. Hasserjian R. Myelodysplastic syndromes and myeloproliferative neoplasms. Part II myelodysplastic syndromes. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=418543043
4. Olga. AML. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=417689596
5. WHO blue book heme 2016.
6. Pozdnyakova O. Myeloid./Lymphoid neoplasms associated with Eosinophilia and rearrangements of PDGFRA, PDGFRB, or FGFR1 or with PCM1-JAK2. Am J Clin Pathol. February 2021; 155:160-178
7. Kelemen K. Eosinophilia / Hypereosinophilia in the Setting of Reactive and Idiopathic Causes, Well-defined Myeloid or Lymphoid Leukemias, or Germline Disorders. Am J Clin Pathol Feb 2021; 155:179-210
8. Hasserjian.Myelodysplastic / Myeloproliferative Neoplasms and Eosinophilic Neoplasms. Society for Hematopathology. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=445215255
9. Prakash. How I Diagnose Primary Myelofibrosis. Am J Clin Pathol 2022; 157:518-530