
Molecular Pathology
Molecular biology
Techniques
Molecular biology
The structure of nucleic acids (DNA and RNA)
Nucleotides
Deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) are chains of nucleotides
- each nucleotide has a pentose sugar, a phosphate group, and a nitrogenous base
- pentose sugars include ribose (for RNA) and deoxyribose (for DNA)
- nitrogenous bases include pyrimidines and purines
-- uracil is unique to RNA, where it substitutes for thymine
-- nomenclature includes the name of the nitrogenous base followed by triphosphate. For example dATP is adenosine triphosphate
- the phosphodiester bond links nucleotides bwt the 5' triphosphate of one nucleotide and the 3' hydroxyl group of another
- polymerization is directional from 5' to 3'
Double stranded DNA
- structure relies upon base pairing through hydrogen bonding of complementary bases
-- cytosine / guanine base pairs share 3 hydrogen bonds; adenine / thymine pairs share 2
- the melting temperature of a double stranded segment - the temperature of a double stranded segment - the temperature at which 50% of the sequence is single stranded - is higher for equivalent length cytosine / guanine rich sequences than adenine / thymine rich sequences
- double stranded DNA is "antiparallel": 1 strand in the 5' to 3' direction base pairs with the other strand arranged in a 3' to 5' direction
- in B form (the MC physiological form), dsDNA measures 2 nm across c 10 nucleotides per complete helical turn, each turn measures 3.4 nm in length, and 2 grooves are formed - major or minor
RNA
Uracil instead of thymine, ribose instead of deoxyribose
- less stable than DNA and has a relatively shorter halflife
- ribosomal RNA (rRNA) plays a structural and catalytic role in ribosomesmessenger RNA (mRNA) translates information in DNA into amino acid sequences (proteins)
- microRNAs (miRNAa) are short fragments of RNA that regulate mRNA levels and consequently protein expression; heterogeneous nuclear RNAs (hnRNAs) include miRNA and mRNA
- transfer RNA (tRNA) delivers specific amino acids to the ribosome for addition to a growing peptide chain; nomenclature is based upon whether amino acid bound (nonacylated/acylated) and particular amino acid, eg tRNA:Ala (nonacylated)/alanyl tRNA, Ala tRNA, or Ala tRNA:Ala (acylated)
Nucleic acid modifying enzymes
Polymerase
catalyzes formation of a phosphodiester bond in 5' tp 3' direction
- subtypesL DNA polymerase; RNA polymerase I produces ribosomal RNA (tRNA); RNA polymerase II produces messenger RNAs (mRNAs) and microRNAs (miRNAs); RNA polymerase III produces the transfer RNAs (tRNAs)
Ligase
- catalyzes the formation of the phosphodiester bonds bwt adjacent ends of nucleotide chains
Nuclease
- catalyzes cleavage of phosphodiester bonds
- 2 main types:
1) endonucleases can cleave within nuclei acid chains; rstriction endonucleases cut in a palindromic seuence specific fashion
2) exonucleases require a free end
Mitochondrial DNA
Mitochondrial genome
Composed of double stranded DNA molecules, one or more per mitochondrion
- 37 genes that encode components of the oxidative phosphorylation complex, tRNAs and rRNAs used in translation of mitochondrial genes
- other mitochondrial proteins are encoded in the nuclear genome
- replication is independent of nuclear genome
Heteroplasmy / homoplasmy
- mitochondrial genomes can undergo mutation; furthermore, when cell divides, daughter cells may end i[ with an uneven distribution of the mitochondria
- the combination of mutation and uneven distribution may lead to heteroplasmy, or nonhomogenous distribution of mitochondrial alleles
- all mitochondria in a cell being genetically identical is referred to as homoplasmy; genetic heterogeneity is referred to as heteroplasmy
- heteroplasmy contributes to variable penetrance of mitochondrial genetic disorders
Maternal inheritance
the mitochondria in an embryo comes exclusively from the egg
- disorders are inherited from the mother's side
Control of gene expression
Epigenetic regulation
Methylation of nucleotides affects cytosine moieties located within cytosine-phosphate-guanine (CpG) islands; this results in decreased DNA transcription in adjacent genes, and the degree of silencing correlates with the number of methylated nucleotides. Forms the bases of imprinting
- histone acetylation "loosens" the histone resulting in increaesd DNA transcription; deacetylation has the opposite effect
Alternate mRNA splicing
- small nuclear ribonucleoproteins or snRNPs form the subunits of the spliceosome complex
- use of different splice acceptor sites is the basis of alternative splicing
- mutations in the splice acceptor, splice donor, or branch site can lead to aberrant splicing
Control of mRNA translation
- mediated by microRNAs (miRNAs), a member of a family of short interfering RNAs, form complementary antisense base pairs with nascent RNA transcripts
Control of the rate of protein degradation
Protein structure
Primary structure
- the linear sequence of amino acids
- the peptide bond links adjacent amino acids; carboxyl group of the preceding amino amino acid bonded to amino group of the subsequent amino acid
Secondary structure
- the way the polypeptide coils
- MC structures are the a helix and the B strand (B pleated sheet)
Tertiary and Quaternary structure
- 3 dimensional structures that form within a polypeptide (tertiary) or bwt polypeptides (quaternary)
DNA replication and cell division
DNA replication
occurs during the S phase of the cell cycle
- is "semiconservative"
- DNA helicase and DNA topoisomerase unwind the double helix
- replication occurs simultaneously at multiple sites (AT rich "origins"), resulting in several replication bubbles
- DNA polymerase catalyzes strand sunthesis in the 5' to 3' direction; on 1 strand the cDNA is formed in uninterrupted fashion (the "leading" strand), while on the other multiple primers and a discontinuous production of cDNA (Okazaki fragments) are formed (the "lagging" strand)
- RNA primers are removed by RNase and DNA is synthesized in the gaps followed by ligation (ligase)
Cell cycle
- begins when cell enters the G1 phase with a diploid (2N) number of chromosomes
- then enters synthesis (S) phase where the chromosomes multiple
- brief G2 phase followed with tetraploid (4N) chromosome complement
- Mitosis (M)
-- Prophase: centrioles move to opposite poles in the cell and the microtubule apparatus begins to form
- Metaphase: nuclear envelope disappears and chromosomes begin to condense and align with the central metaphase plate
- anaphase: separation of sister chromatids (duplicated chromosomes)
- telophase
Terminally differentiated cells are in a phase referred to as G0
Meiosis occurs only in the germ cells
- begins c diploid cell
- DNA replication raises the DNA content from 2N to 4N; 4 chromatids composed of 2 copies each of maternal and paternal chromosomes, joined at centromere
- enters meiosis I: recombination occurs in prophase I at synaptic connections; in oocytes the process pauses after prophase I (dicytotene stage), during adulthood completes meiosis I and pauses again at metaphase II unless fertilized
- nondisjunction most commonly occurs during meiosis I, but can also occur during meiosis II and results in aneuploidy
Genetic anomalies
Mutation vs polymorphism
- mutation: genetic change often c a potential deleterious effect
- polymorphism: not deleterious and present in at least 1% of the population
Mutations may be classified according to effect on protein function
- loss of function or gain of function
Mutations may be classified according to effect on gene structure
- point mutations, insertion mutations, deletion mutations, inversions, translocations
- point mutations further subcategorized according to their effect
- nonsense mutations result in premature truncation of translation
- missense mutations lead to changes in amino acid sequence
- silent mutations change the nucleic acid sequence but do not result in the productions of either a stop codon or different amino acid
- splice mutations affect splicing donor or acceptor sequences
- frameshift mutations lead to a change in the reading frame of the ribosome translation
Chromosomes
Nucleosome structure
- octamer of positively charged histone proteins, 2 each of H2A, H2B, H3, H4
- wrapped around this is 146 bases of DNA (which has negative charge)
- between adjacent nucleosomes is a short linker of ~20-50 bases
- each nucleosome is linked to the next by the linker histone, H1
- nucleosomes are stacked into the 30 nm chromatin fiber c 6 nucleosomes per turn; 50x more compaction accomplished c looping; 20x more with loops into minibands; end result is the chromosome
Chromosome nomenclature
- numbered according to the length of each chromosome, from longest (1) to shortest (22, which actually is not the shortest; that's 21); and is overall based on the International System for Human Cytogenetic Nomenclature (ISCN)
Chromosomes classified by the location of the centromere, and also previously into groups A-G based on shape and size, and another group for the sex chromosomes
- metacentric chromosomes have 2 arms of roughly the same length
- submetacentric chromosomes have a short (p) arm and a long (q) arm
- arcocentric chromosomes have virtually no p arms; the short areas in place of p arms encode the majority of rRNAs and are frequent sites of translocation
Regions of the arms numbered in relation to the centromere; nearest region labeled "l"; each region is further divided into bands and, possibly, subbands. The nomenclature used is [chromosome][arm][region][bamd][subband]
- bands are parts of a chromosome that are clearly distinguishable from adjacent segments by appearing lighter or darker (sub-bands apparent c inc resolution)
- chromosomes are also broken down into regions based on landmarks (such as the centromere, telomere, and bands
- centromeres are termed cen, and the short and long terminals are pter and qter
In describing structural changes, there is a short form that defines bands where chromosomal breakage occurs, and a long (detailed) form that identified the abnormality by describing the entire structure of an altered chromosome
Nomenclature examples
Polyploid
Techniques
Cytogenetics
Karyotyping
- dividing cells arrested in metaphase
- staining c Giemsa (G banding), quinacrine (Q banding), or a number of techniques to produce a reverse staining pattern to G banding (R banding)
- G banding stains A-T rich areas more intensely than G-C rich areas
Molecular
Resources
ClinVar
https://www.ncbi.nlm.nih.gov/clinvar/
- aggregates info on genomic variation and relationship to human health
PolyPhen-2
http://genetics.bwh.harvard.edu/pph2/
- predicts possible impact of an aa substitution on structure/function of protein using physical and comparative considerations
Isolation of nucleic acid
- DNA extraction involves disrupting membranes, degrading or precipitating proteins, and centrifucation or extraction to separate the nucleic acid in aqeous phase from membranes/protein, the nucleic acid can then be isolated from aqueous phase by alcohol precipitation and dehydration; RNA is less stable than DNA
- extraction verified through spectrophotometric absorption at 260 nm; protein absorbs light maximally at 280 nm, so that purity of DNA can be assessed c a ratio of absorbance at 260 nm to 280 nm; ratio <1.8 is considered to be relatively pure
DNA amplification by PCR
thermostable polymerase, such as from Thermus aquaticus (Taq) is used along with template DNA, primers, deoxynucleotides, disvalent cation, and pH buffer
- single cycle involves denaturation at 95 C; annealing at lower temperature (dependent on primer and sequence variables); temperature raised to 72 C for polymerization; return to 95 C to begin next cycle
- amount of DNA produced under optimal conditions: [DNA] = 2^n where n= # cycles. A 10 fold amplification takes place every 3.3 cycles (2^3.3=10); if there are 100 copies in cycle 7 then there will be 1000 copies in cycle 10
- after 30-40 cycles the reaction products may be analyzed by agarose gel electrophoresis and ethidium bromide staining
Methylation specific PCR
- to detect methylation of genes
- methylated cytosine residues in the presence of sodium metabisulfite are reduced to uracil
- after pretreatment with metabisulfite, PCR primers that are specific to a sequence containing uracils are utilized to selectively amplify the methylated sequence
Reverse transcriptase PCR
- to detect specific RNA sequences
- reverse transcriptase used to make DNA copy of RNA
- subsequently a 2nd DNA strand is synthesized from the 1st cDNA strand and a double stranded DNA representation of the RNA is produced; this dsDNA can then be used as a template for PCR
Real time PCR
- a fluorescent dye is used to detect cDNA as it formsl the cycle number at which the increase in fluorescence is exponential is directly related to the starting DNA
- a horizontal line (known as the threshold) is set by the user; the point at which the fluorescence crosses the threshold (enters the logarithmic phase) is called the Ct
- a sample that has a quantity, X, of template would cross this threshold 1 cycle later than a sample c 2x template; samples that differ by a factor of 10 would be ~3.3 cycles apart
Melting point analysis
- to aid in identification of the PCR product
- following PCT, using nonhyrolyzable probes or intercalating dye, fluorescence measured while incrementally increasing the temperature
- the melting [point is the point at which 50% of the DNA is single stranded; on a plot of fluorescence vs temperature the melting point is the point of the maximal change in rate of melting
- melting point is dependent on length, G:C content and amount of mismatch
Multiplex PCR
- allows for the identification of multiple PCR products at the same time
- multiple templates and primers are added to the mix and run in one tube at the same time
- can have issues with competition for limited resources within reaction tube
Transcription mediated amplification (TMA) and nuclei acid sequence based amplification (NASBA)
- for isothermal amplification of RNA targets; primarily used for infectious disease applications
- utilize specific sequences that are recognized by bacteriophage RNA polymerase in order to make more RNA copies
- RNA copies produced can then be probed
Multiplex ligation dependent probe amplification (MLPA)
- to interrogate multiple mutations at the same time
- utilizes numerous specific probes varying in the length of an extragenetic stuffer sequence; each probe contains gene specific sequence and sequence shared c all the other probes used
- for each possible mutation there is a specific probe and an adjacent anchor probe separated by a single base
- only when the 2 probes anneal adjacent to each other are they able to be subsequently ligated and then amplified with probe specific primers
- can be adapted for determination of single nucleotide polymorphisms/mutation, dosage and copy number, and methylation status
Signal amplification
- techniques that amplify signal rather than amplifying DNA
- less sysceptible to contamination but not as sensitive as target amplification
- can amplify the probe (the cleavase reaction or ligation amplification reaction) or the signal (the branched DNA or hybrid capture techniques)
Restriction endonucleases
- named according the bacterial species from which they were isolated (HindIII from H influenza, EcoRI from E coli)
- sequence specific cleavage activity such that cleave DNA in predictable manner
- mutations can result in either loss or gain of a recognition sequence
- exploited in Southern blot or the restriction fragment length polymorphism (RFLP) assay
- methylation status can be deduced from whether the sequence is protected from endonuclease digestion
Blotting (Southern, northern, western)
- DNA is Southern blotted, RNA is northern blotted, and protein is western blotted
- once affixed to the membrane the nucleic acid or protein can be probed to characterize
DNA sequencing
- Sanger dideoxy sequencing
= based upon termination of strand elongation when a dideoxy base is incorporated (lacks 3' end), resulting in a mixture of strands of varying length
- this mixture subjected to electrophoresis, resulting in a ladder, each ring representing an aborted replication due to incorporation of the selected dideoxy chain terminator base
- if 4 such mixtures are obtained, 1 for each base, then the sequence can be determined
- modern techniques utilize a single reaction with differentially-colored fluorochrome labeled dideoxy bases subjected to capillary electrophoresis and fluorescent detection
Pyrosequencing (single base extension)
- quantitative measurement of the pyrophosphate is released in a stoichiometric amount in relation to the number of bases incorporated, the sequence of a target DNA can be inferred
Hybridization techniques (FISH, CGH)
Probes
Chromosome enumeration probes (CEP) are targeted to conserved regions near centromeres
- locus specific probes - targeted to a particular dequence
-- fusion probes are useful for well defined translocations with conserved break/ fusion points
-- breakapart probes useful when gene may be useful when translocated with a variety of partners
- Whole chromosome paint probes - a number of probes designed to provide full coverage of a chromosome
Comparative genomic hybridization (CGH)
Conventional CGH: variety of probes are applied to metaphase chromosomes for determination of chromosomal / subchromosome copy number; commonly used for genetic characterization of chromosomal anomalies in children and in tumor cells; not able to detect balanced translocations; only unbalanced abnormalities
Array CGH: for detection of chromosomal copy number changes on a genome wide scale, often used in the workup of a mentally / developmentally delayed patient with a normal appearing karyotype; provides higher resolution than conventional CGH
Applications
Assessment of short tandem repeats
Short tandem repeats (STRs) are runs of repeated oligonucleotides (2-5) that are normally present in the genome
- length (copy number) of STRs is stably inherited and is normally stable from cell to cell, but different from person to person
- uses include determination of parentage, identification of remains for forensic purposes and assessment of chimerism in transplant recipients
- in abnormal conditions, STRs are unstably inherited (trinucleotide repeat disorders) or unstable within an individual (mismatch repair disorders)
Assessment of single nucleotide polymorphisms
- single nucleotide polymorphisms (SNPs) are single base pair differences bwt individuals
- present in at least 1% of the population and do not cause disease
- present on the average of 1 for every 1,000 bases
- grand total of an individual's SNP is referred to as the haplotype
- used in: DNA fingerprinting for forensic purposes; SNPs vary geographically and can be used for forming genetic family tree; SNPs influence predisposition to certain diseases, traits, or responses to medications
Whole genome sequencing
- Next generation sequencing or high throughput sequencing refers to techniques used to elucidate large genomic sequences
- there are now several different platforms that are capable of sequencing entire genomes
- involves massively parallel sequencing followed by computer-mediated contiguous sequence alignment
- variation includes whole exome sequencing, or selective sequencing of exons
Pharmacogenomics
Cytochrome P450 (CYP) family
- nomenclature supergene family (CYP), family, subfamily, isoenzyme, allele variant, eg the wild type (*1) allele of the 2 family, D subfamily, 6 isozyme is transcribed as CYP2D6*1
- polymorphisms in CYP affect the inactivation and clearance of drugs as well as the activation of a prodrug
- examples:
-- CYP2D6 affects metabolism of codeine to the active metabolite morphine
-- CYP2D6 also have a role in the metabolism of tricyclic antidepressants, such as nortryptyline
- CYP2C9 plays an important role in the metabolism of warfarin and phenytoin
- CYP2C19 responsible for the metabolism of omeprazole, phenytoin, and diazepam
- Vitamin K epoxide reductase (VKORC1) and is involved in the metabolism of vitamin K and inhibited by warfarin
- N-acetyltransferase is central to the metabolism of isoniazid, some polymorphisms result in increased or decreased activity
Next Generation Sequencing (NGS)
aka Massively Parallel Sequencing
- simultaneous sequencing of millions of fragments of DNA (or complementary DNA)
Tests for germline (inherited) and somatic (acquired) mutations
- germline diseases can have a targeted panel, whole exome, whole genome, or mitochondrial DNA sequencing
-- whole exome sequencing usually done when targeted panels are uninformative, and requires testing the child and both parents (trio testing)
-- NGS can be used to analyze cell-free DNA prenatally
-- targeted panels can be broad or focused, and individual genes can be completely or partially sequenced
--- whole exome and whole genome sequencing not currently used for clinical oncology testing
- NGS can be used for circulating tumor DNA tests, HLA, microbial analysis, RNA sequencing, methylation
HLA typing with NGS was difficult initially due to problems differentiating low frequency alleles from high-freq artifacts and distinguishing 2 similar alleles as distinct
- stepwise threshold clustering has helped with these problems
- short tandem repeats (STRs) regions and other tandem repeat regions were problematic
- circulating tumor DNA (ctDNA) now being used clinically, and is referred to as a liquid biopsy
-- mutation detection in ctDNA has helped to guide tx in pts for tyrosine kinase inhibitor response with EGFR activating mutations in lung cancer
-- testing for ctDNA has a lower sensitivity than testing on tumor tissue itself
- high false positive and false negative rates limit practical use of ctDNA for screeining for early cancer dx
Wet Bench Steps
samples undergo DNA extraction, library preparation, target enrichment and sequencing
DNA extraction, almost all methods are acceptable, such as from FFPE
- DNA quantification done by Qubit, or Picogreen
Library Preparation: preparing DNA for use on a sequencer; breaks DNA into fragments and adds adaptors to the ends
- adaptors can include molecular bar codes (to allow pooling of pt samples), universal PCR primers, hybridization sequences to bind DNA fragment to surface, and recognition sites to initiate sequencing
- libraries are fragments of DNA c flanking adaptors that are ready for sequencing
- insert size is the size of the DNA fragment bwt the adaptors
-- short and long fragments have different advantages (short fragments more likely to fall in an exon which is usually the area of interest, and long ones more likely to be in an intron and can help find structural rearrangements)
Target Enrichment - resulting library undergoes enrichment for both whole exome analysis and targeted testing or is sequenced directly for whole genome analysis
- can be performed by hybridization to complementary sequences (sequence capture) or by PCR (usually combined with library preparation step bc primers selecting region of interest also have adaptor sequences)
- sequence capture preferred for larger genomic regions and PCR for smaller regions
Sequencing done on one of two types of instrument:
1) Illumina (HiSeq, MiSeq, NexSeq)
- first step is clonal amplification using a flow cell that hybridizes to part of the adaptor on DNA fragments
- use sequencing by synthesis c fluorescent detection; all 4 fluorescently tagged nucleotides added and compete for the next space
- complementary tagged nucleotide binds but a blocker prevents addition of >1 nucleotide per round (reversible terminator chemistry) c remaining unbound nucleotides washed away
- laser excitation leads to a fluorescent emission that is recorded (simultaneously for each DNA fragment cluster)
- fluorescent tag and blocker are cleaved and next round begins
- per round, 1 base pair can be repeated on the opposite end of the DNA fragment, termed paired end reads
2) Ion Torrent series (IonPGM, IonProton, IonS5)
- first step is clonal amplification using bead emulsion that hybridizes to part of the adaptor on DNA fragments
- beads are then placed in a well (1 bead per well)
Input DNA concentration critical to ensure only 1 DNA fragment binds per bead and ensures DNA fragments well spread out on flow cell
- clonal amplification step creates a bead or cluster c aprrox 1000 identical copies of a unique parent DNA molecule that are physically isolated from other molecules
- only a single base added each round, with a hydrogen ion released when an added base is incorporated which causes a pH change detected for each bead in a well (if a base is not incorporated no voltage is generated)
-- if >6-8 bases are incorporated the signal is no longer proportional and the exact amount cannot be determined
Bioinformatics
Raw data reads for either instrument type undergoes a series of bioinformatics processes (called the pipeline) to deliver a variant call file (VCF)
- processes include demultiplexing, quality analysis, resequencing (mapping reads to a reference genome), and variant identification / annotation
Bar codes to tag DNA fragments of specimen allows multiple samples to be pooled and run together, which decreases sequencing cost
- requires a demultiplexing step where reads are sorted by bar code/ sample for further analysis
- the demultiplexed file c raw reads call the FASTQ file
After demultiplexing, individual reads for a sample are mapped to a reference genome (BAM file) and differences bwt the two are compared
- identical (duplicate) reads discorded for whole genome sequencing or sequence capture but not for amplicon-based sequencing
-- if multiple reads show the same difference, a variance is called (threshold for number of reads is determined by the lab and should be validated)
- a heterozygous single nucleotide variant (SNV) should be present in 50% of reads, though can range from 23-74% in real practice
- output file defines all variants for a sample and their allelic fractions in a variant call file
-- the list of variants undergoes interpretation
- any changes from the wet bench to the informatics portions of the test require revalidation
Interpretation of Variants
Complex when involving whole genes
- the greater the size of the genome sequenced, the larger the probability of finding a rare or novel variant
Must be able to mask for certain diseases that the patient may not want to know (ie some late onset adult dz with no tx) while also making available certain things that may be helpful (such as drug metabolism genes if needed)
Validation
Must be validated throughout the entire process (from DNA extraction to the bioinformatics pipeline)
- validation process shows ability to find genetic changes such as single nucleotide changes, indels
- sensitivity, specificity and reproducibility should be established for all assays
- should document areas that are not able to be sequenced
- changes only in the bioinformatics pipeline can be compared using the new and old processes
- changes in the wet bench process require end to end revalidation, but can use fewer samples than the original validation
Cost
Based on 1) library preparation (reagents, labor, necessary equipment, 2) selection strategy (PCR or capture) and 3) the sequencer used
- costs vary widely depending on the method used
- library preparation is combined with selection for PCR-based methods, which reduces the combined cost of these 2 steps
- cost also related to how much of the sequencer capacity is used for a sample and whether the sequencer is at full capacity on a given run
In general, the higher the number of samples included in an analytic run, the lower the per-sample cost
Limitations
Analytic Sensitivity
Sensitivity for NGS for SNV detection is 5-10%
- this is a limitation in minimal residual disease, with low tumor percentages
-- this low sensitivity may be due to PCR noise in addition to C to T transversions in FFPE tissue, sequencing errors and systematic errors
---FFPE has higher artifacts than fresh tissue samples
-- counterintuitively, the error rate is higher c increasing coverage
- neither PCR nor fixation causes insertions / deletions (indels), and outside of repeat regions there is better sensitivity for detecting small indels than SNVs
How to improve sensitivity?
1) Use of overlapping paired end reads
- DNA insert size must be the same size or smaller than the number of reads
2) use random nucleotide tags, aka unique identifiers (UIDs) or primer IDs, as they are often incorporated into the PCR primer
- random nucleotide tags added to DNA fragments, assigned if DNA was sheared, or incorporated during the first round or two of PCR for amplicon-based methods
-- these steps occur before amplification and result in a DNA fragment c a random and unique nucleotide sequence at one or both ends
- all reads that map to the same location and have an identical UID are considered part of a UID family and will be analyzed as a group
- if a mutation is present in a majority of that UID family, the mutation is considered present and is considered 1 read
Difficult to Sequence Areas
Homologous regions, repetitive regions, and GC-rich areas not reliably interpretable by current NGS and standard bioinformatics algorithms
- homologous regions (including pseudogenes) are areas of genome c high sequence similarity that can differ from gene of interest by only a few base pairs
- DNA fragments sequenced from target gene and homologous regions can be so similar that they may be indistinguishable; inc risk of this happening c shorter sequences
- reads from a homolog can be mismapped to a real gene and vice versa, leading to inaccurate results
- newer machines with longer read segments may solve this
- in repeat areas, need a unique flanking sequence to reliable map a sequence read and determine repeat size
- stutter (polymerase slippage leading to small shifts in repeat size) and PCR sequencing mistakes can be sources of error
- GC-rich regions have high background noise and lower sequence quality, esp c Illumina sequences which give substitution errors
- should document these difficult to sequence areas during validation
Limitations in Databases and Knowledge
Layer 1 databases (aka Clinical Genomic Variant Repository) have only sequence / variant information
Layer 2 database (aka Genomic Medical Data Repository) have sequence / variant information with clinical / phenotype data
Layer 3 databases (Genomic Medical Evidence Database) have medical evidence of classification or assoc c sequence / variant information
Only ClinVar and dbSNP have data for inherited diseases and somatic mutations
Structural Variation and Copy Number Variation
NGS not able to detect structural rearrangements or copy number variations (CNVs), though is good at detecting SNVs and small inserion / deletions (indels)
- techniques being used to detect CNVs include depth of coverage (read depth), read pair, split pair, assembly based, or a combination of these techniques
- parts of the genome are more prone to false-positives than others
Read pair (mate pair) analysis compares distance of 2 ends of a read pair to the avg insert size
- requires oaured end reads, only detects smaller CNVs, and is limited by the insert size, and only detects smaller CNVs
- an advantage is that it detects CNVs and rearrangements
Split pair (split read) analysis looks specifically at paired reads, where one of the paired reads fails to map or maps only partially
- also requires paired end reads, only detects smaller CNVs and does not perform well in regions of low complexity
- it can pinpoint breakpoints and detect rearrangements
Assembly-based analysis uses de novo alignment of reads, which matches the individual reads to each other instead of a reference genome
- is computationally more intense and works best on smaller genomes (such as bacteria) but can be used clinically

A, Overview of wet bench steps for capture-based sequencing. DNA undergoes library preparation followed by capturebased selection before sequencing. B, Overview of wet bench steps for polymerase chain reaction (PCR)–based sequencing. The PCR selection step occurs before library preparation or may be combined with the librarypreparation step in PCR-based sequencing.


Short insert size
Long insert size
Fragments with a short DNA insert size (top) are more likely to have both paired end reads (red bars) fall within the exon. Fragments with a long insert size are more likely to span the breakpoint of a rearrangement, which often occurs in the intron. Reprinted from Yohe SL. Hot topic spotlight—new frontiers in clinical next-generation sequencing. In: Cushman-Vokoun AM, Anderson WB, eds. Precision Medicine Resource Guide. Northfield, IL: College of American Pathologists; 2016:12. With permission from College of American Pathologists. Copyright 2016


Illustration of Illumina sequencing by synthesis (Athrough D) and Ion Torrent ion-based sequencing (E). A, Fluorescently tagged nucleotides (blackcircles with colored circles) compete for the next complementary space on the DNA strand (gray circles). B,When a fluorescently tagged nucleotide is incorporated, it blocks further addition of nucleotides. C, The flowcell is washed, removing additional fluorescently tagged nucleotides, and a laser signal leads to fluorescent emission. D, The fluorescent tag and blocker are removed and washed away, allowing incorporation of the next base during the next cycle. This occurs simultaneously for all DNA strands in a cluster and all clusters on the flowcell. E, In each cycle, a single base is added in a set pattern. For this example, the order of base additions is A, T, C, and G, which then repeats. If a base is incorporated an ion is released, leading to a pH(voltage) change that is proportional to the number of bases added in a row. Reprinted from Yohe SL. Hot topic spotlight—new frontiers in clinical next-generation sequencing. In: Cushman-Vokoun AM, AndersonWB, eds. Precision Medicine Resource Guide. Northfield, IL: College of American Pathologists; 2016:13–14. With permission from College of American Pathologists. Copyright 2016.


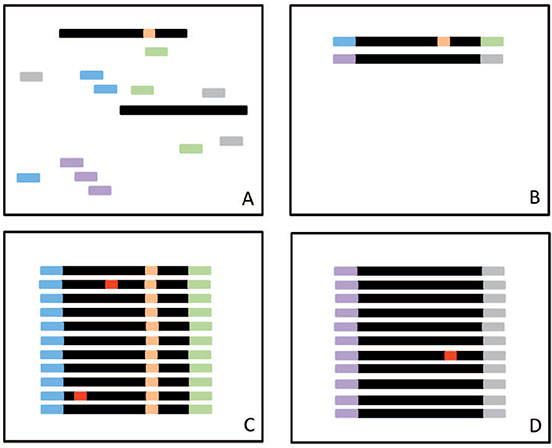
A, Before amplification, random tags (short bars) are added to DNA fragments (black), some of which have a mutation (orange). B, The tags randomly attach to DNA fragments. C, During amplification some copies will develop an error (red). All fragments will be sequenced. Only mutations that are detected within a majority (eg, 95%) of all the sequencing reads with the same ID tag will be identified as true mutations. D, Mutations present in a minority of reads with the same ID tag are considered errors. Reprinted from Yohe SL. Hot topic spotlight— new frontiers in clinical next-generation sequencing. In: Cushman-Vokoun AM, Anderson WB, eds. Precision Medicine Resource Guide. Northfield, IL: College of American Pathologists; 2016:5. With permission
from College of American Pathologists. Copyright 2016.

The right side shows the CYP21A2 gene with baits (Table 2) designed for sequence capture (green bars). The left side shows the CYP21A2 pseudogene without baits. A similar number of sequencing reads are being mapped to the pseudogene as to the real gene; since the reads are so similar the actual source cannot be determined. These reads would have a low mapping quality score, as the reads are mapping to more than 1
location, as indicated by the faded colors. Black arrow: coverage (gray peaks), green circle: location of baits (if any). Cropped integrated genome viewer (IGV) screenshot (Broad Institute, Cambridge, Massachusetts). 121, 122 Reprinted from Yohe SL. Hot topic spotlight—new frontiers in clinical next-generation sequencing. In: Cushman-Vokoun AM, Anderson WB, eds. Precision Medicine Resource Guide. Northfield, IL: College of American Pathologists; 2016:6. With permission from College of American Pathologists. Copyright 2016.
References
1. Yohe S, Thyagarjan B. Review of Clinical Next-Generation Sequencing. Arch Pathol Lab Med. 2017;141:1544–1557; doi:
10.5858/arpa.2016-0501-RA.