
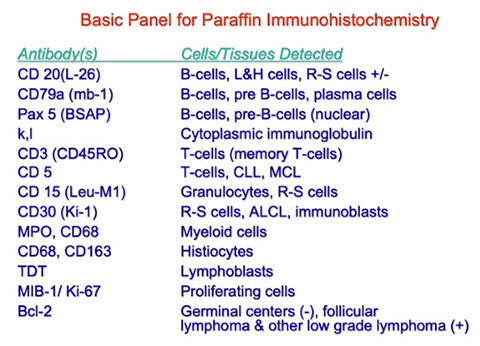
Lymphoid Neoplasms
Lymphocyte development
Reactuve lymphocytosis
Precursor lymphoid neoplasms
Mature T- and NK-cell neoplasms
B lymphoblastic leukemia / lymphoma, NOS
B lymphoblastic leukemia / lymphoma with recurrent genetic abnormalities
- B-lymphoblastic leukaemia/lymphoma with t(9;22)(q34.1 ;q11 2); BCR-ABL1
- B-lymphoblastic leukaemia/lymphoma with t(v; 11 q23.3); KMT2A-rearranged
- B-lymphoblastic leukaemia/lymphoma with t(12;21)(p13.2;q22.1); ETV6-RUNX1
- B-lymphoblastic leukaemia/lymphoma with hyperdiploidy
- B-lymphoblastic leukaemia/lymphoma with hypodiploidy
- B-lymphoblastic leukaemia/lymphoma with t(5;1 4)(q31.1;q32. 1); IGH/IL3
- B-lymphoblastic leukaemia/lymphoma with t(1; 19)( q23;p 13.3); TCF3-PBX1
- B-lymphoblastic leukaemia/lymphoma, BCR-ABL1-like
- B-lymphoblastic leukaemia/lymphoma with iAMP21
T-lymphoblastic leukaemia/lymphoma
- Early T-cell precursor lymphoblastic leukaemia
- Indolent T-lymphoblastic proliferation
NK-lymphoblastic leukaemia/lymphoma
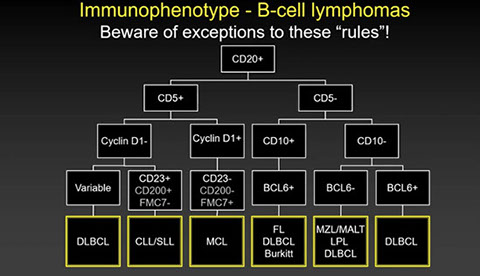
Mature B-cell neoplasms
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL)
- Monoclonal B-cell lymphocytosis
B-cell prolymphocytic leukemia (PLL)
Splenic marginal zone lymphoma (sMZL) - see Spleen and LN
Hairy cell leukemia (HCL)
Splenic B-cell lymphoma/leukemia, unclassifiable
- Splenic diffuse red pulp small B-cell lymphoma
- Hairy cell leukaemia variant
Lymphoplasmacytic lymphoma
lgM Monoclonal gammopathy of undetermined significance
Heavy Chain Diseases (HCD)
- Mu heavy chain disease
- Gamma heavy chain disease
- Alpha heavy chain disease
Plasma cell neoplasms
- Non-lgM monoclonal gammopathy of undetermined
significance
- Plasma cell myeloma (MM)
- Plasma cell myeloma variants
-
- Smoldering (asymptomatic) plasma cell myeloma
- Non-secretory myeloma
- Plasma cell leukemia
- Plasmacytoma
-
- solitary plasmacytoma of bone
- Extraosseous [extramedullary] plasmacytoma
- Monoclonal immunoglobulin deposition diseases
-
- Primary amyloidosis
- Systemic LC and HC deposition dz's
- Plasma cell neoplasms with associated paraneoplastic syndrome
-
- Osteosclerotic myeloma (POEMS syndrome)
- TEMPI syndrome
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma)
Nodal marginal zone lymphoma (NMZL)
- Pediatric nodal marginal zone lymphoma (PNMZL)
Follicular lymphoma (FL)
- Testicular FL
- In situ follicular neoplasias
- Duodenal-type FL
Pediatric-type follicular lymphoma
Large B-cell lymphoma with IRF4 rearrangement
Primary cutaneous follicle center lymphoma
Mantle cell lymphoma (MCL)
- Leukemic non-nodal mantle cell lymphoma
- In situ mantle cell neoplasia (ISMCN)
Diffuse large B-cell lymphoma (DLBCL), NOS
T-cell/histiocyte-rich large B-cell lymphoma (TCHRLBCL)
Primary diffuse large B-cell lymphoma of the CNS
Primary cutaneous diffuse large B-cell lymphoma, leg type
EBV-positive diffuse large B-cell lymphoma, NOS
EBV-positive mucocutaneous ulcer
DLBCL associated with chronic inflammation
- Fibrin-associated diffuse large B-cell lymphoma
Lymphomatoid granulomatosis (LYG)
Primary mediastinal (thymic) large B-cell lymphoma (PMBL)
Intravascular large B-cell lymphoma
ALK positive large B-cell lymphoma
Plasmablastic lymphoma (PBL)
Primary effusion lymphoma (PEL)
HHV8-associated lymphoproliferative disorders
- Multicentric Castleman disease (see Spleen and Lymph Node)
- HHV8-positive diffuse large B-cell lymphoma, NOS
- HHV8-positive germinotropic lymphoproliferative disorder
Burkitt lymphoma
Burkitt-like lymphoma with 11 q aberration
High-grade B-cell lymphoma
- High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements
- High-grade B-cell lymphoma, NOS
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classic Hodgkin lymphoma
T- cell prolymphocytic leukemia (T-PLL)
T-cell large granular lymphocytic leukemia (LGL leukemia)
Chronic lymphoproliferative disorder of NK cells
Agressive NK cell leukemia (ANKL)
Ebstein-Barr virus (EBV) positive T-cell lymphoproliferative diseases of childhood
- Systemic EBV+ T-cell lymphoma of childhood
- Chronic active EBV infection of T- and NK-cell type, systemic form
- Hydroa vacciniforme-like lymphoproliferative disorder
- Severe mosquito bite allergy
Adult T-cell leukemia / lymphoma (ATCL/ATLL)
Extranodal NK-T-cell lymphoma, nasal type (NTNKT)
Intestinal T-cell lymphoma
- Enteropathy-assoc T-cell lymphoma (EATCL)
- Monomorphic epitheliotropic intestinal T-cell lymphoma
- Intestinal T-cell lymphoma, NOS
- Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract
Hepatosplenic T-cell lymphoma (HSTL/HSTCL)
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL)
Mycosis fungoides (MF)
Sezary syndrome
Primary cutaneous CD30 positive T-cell lymphoproliferative disorders
- Lymphomatoid papulosis (see Skin)
- Primary cutaneous anaplastic large cell lymphoma
Primary cutaneous peripheral T-cell lymphomas, rare subtypes
- Primary cutaneous gamma-delta T-cell lymphoma
- Primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma
- Primary cutaneous acral CD8-positive T-cell lymphoma
- Primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder
- Peripheral T-cell lymphoma, NOS (PTCL)
Angioimmunoblastic T-cell lymphoma and other nodal lymphomas of T follicular helper (TFH) cell origin
- Angioimmunoblastic T-cell lymphoma
- Follicular T-cell lymphoma
- Nodal peripheral T-cell lymphoma with TFH phenotype
Anaplastic large cell lymphoma (ALCL), ALK positive
Anaplastic large cell lymphoma (ALCL), ALK negative
Breast implant-associated anaplastic large cell lymphoma
Hodgkin lymphomas
Hodgkin Lymphoma Overview
Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL)
Classic Hodgkin Lymphoma (cHL)
- Nodular sclerosis classic Hodgkin lymphoma
- Mixed-cellularity classic Hodgkin lymphoma
- Lymphocyte-rich classic Hodgkin lymphoma
- Lymphocyte depleted classic Hodgkin lymphoma
Immunodeficiency-associated lymphoproliferative disorders
Lymphoproliferative diseases associated with primary immune disorders
Lymphomas associated with HIV infection
Post-Transplant Lymphoproliferative disorders (PTLD)
- Non-destructive PTLD
- Polymorphic PTLD
- Monomorphic PTLD (B- and T/NK-cell types)
-
- Monomorphic B-cell PTLD
- Monomorphic T/NK-cell PTLD
- Classic Hodgkin lymphoma PTLD
Other iatrogenic immunodeficiency-associated lymphoproliferative disorders
Histiocytic and dendritic cell neoplasms
Hemophagocytic Lymphohistiocytosis, Rosai-Dorfman disease (see Spleen and Lymph Node)
Histiocytic sarcoma
Tumors derived from Langerhans cells
- Langerhans cell histiocytosis (LCH)
- Langerhans cell sarcoma (LCS)
Indeterminate dendritic cell tumor
Interdigitating dendritic cell sarcoma
Follicular dendritic cell sarcoma
- Inflammatory pseudotumour-like follicular/fibroblastic dendritic cell sarcoma
Fibroblastic reticular cell tumor
Disseminated juvenile xanthogranuloma
Erdheim-Chester disease
Lymphocyte Development
~9/10 lymphoid neoplasms are mature B cells, which present as lymphadenopathy usually
- MC B-cell neoplasms = DLBCL> FL >CLL > MCL
- MC B-cell leukemia = CLL
- may also have cytopenia, B sx, and M protein
- if extranodal, MC in stomach
- high grade lesions usually localized, vs low-grade lesions, which are disseminated
- hg in kiddos, l-g in adults; M>F, Fs get PMBL, FL, or MALT lymphoma
- immunodef and autoimmunity are risk factors
- 2/5 in BM at presentation, bilat sampling inc sensitivity by 15%
FL inc BM in up to 2/5, PB in 1/5, discordance in 1/3
- has paratrabecular distribution
DLBCL in BM in 3/20, PB in 1/3, discordance in 1/3
LPL in BM in 1/10, PB in 1/10
- has interstitial pattern
MZL in 1/20 BM, PB in 4/5
Burkitts in BM in 1/2-, PB in 2/5
B- and T-cell neoplasms can be classified to some extent by the stage of their maturation
- some neoplasms don't fit this though, like Hairy Cell Leukemia in B-cell neoplasms
- rarely can show lineage heterogeneity or lineage plasticity
NK cells closely related to and shre immunophenotypic and functional properties with T cells, which is why NK and T cell neoplasms grouped together
Innate Immune System
Provides the first line of defense
- a primitive response, including barrier immunity in mucosal and cutaneous surfaces
-- don't need to encounter antigens like MHC molecules, and don't need antigen-presenting cells to initiate an immune response
- does not result in immunologic memory
- includes NK cells, CD3+ CD56+ T cells (NK-like T-cells), and gamma-delta T-cells
Adaptive Immune System
More sophisticated; 2 key features: specificity and memory
-
B-cells
Neoplasms of B-cells mimics different stages of cell development, which forms most of its classification system
B-lymphoblasts undergo IG VDJ gene rearrangement and differentiate to mature surface immunoglobulin (sIg)-positive (IgM+ IgD+) naive B-cells via pre-B cells c cytoplasmic mu heavy chains and immature IgM+ B cells
Naive B cells, which are usually CD5+, are small resting lymphocytes that circulate in PB and are also found in primary lymphoid follicles and follicle mantle zones (aka recirculating B-cells)
- many cases of mantle cell lymphoma thought to correspond to CD5+ naive B-cells
Naive B-cells can become plasma cells or memory B-cells after encountering antigen
- Naive B-cells can mature directly to plasma cells that make IgM in response to antigen
- other antigen-exposed B-cells migrate to the center of a primary follicle, proliferate and fille the follicular dendritic cell meshwork, forming a germinal center
Germinal center centroblasts have low sIg and switch off BCL2 expression
- GC centroblast progeny can undergo apoptosis
- Centroblasts are CD10+, BCL6+
-- BCL6 is not expressed in naive B-cells and is turned off in memory B-cells and plasma cells
- other germinal center markers recently describes are LMO2 and HGAL
LMO2 found in precursor cells in BM, but seems specific for germinal center B-cells in normal reactive lymphoid tissues
- lacks specificity in lymphoid neoplasms
HGAL (aka GCET2) found in germinal center B-cells and germinal center-derived malignancies
In germinal centers (GC), somatic hypermutation occurs in IGV genes
- can result in non-functional gene or a gene that produces antibody with more or less affinity for an antigen
- also in the GC, some cells switch from IgM to IgG or IgA production
-- this is how the GC reaction gives rise to higher-affinity IgG or IgA antibodies of the late primary or secondary immune response
- BCL6 undergoes somatic mutation in the GC, but at a lower frequency than the IG genes
- ongoing IGV gene mutation with intraclonal diversity is a hallmark of germinal center cells, and both IGV gene and BCL6 mutations are markers that cells have been through the germinal center
-- most DLBCLs somewhat resemble centroblasts with mutated IHV genes, consistent with exposure to the GC
-- Burkitt lymphoma cells also have mutated IGH genes and are BCL6 positive and are throught to correspond to germinal center blasts
Centrocytes, seen in the light zones of GCs, are matured centroblasts
- centrocytes express sIg with an altered antibody-combining site comared to their progenitors, due to somatic mutations and heavy-chain class switching
- centrocytes that have mutations with increased affinity are saved from apoptosis and re-express BCL2
- surface molecules (CD23 and CD40) on follicular dendritic cells and T-cells interact with centrocytes by switching off BCL6 expression, causing them to differentiate to memory B cells or plasma cells
- BCL6 and IRF4 (MUM1) are reciprocally expressed, with IRF4 positivity seen in late centrocytes and plasma cells
- IRF4 (MUM1) plays a critical role in downregulating MUM1
MYC plays important role in germinal center formation
- MYC is upregulated on interaction of naive B cells with antigen and T cells by BCL6
- in normal reactive lymph nodes, MYC highlights some centrocytes in the light zone of the GC and is repressed in the dark zone
- however, MYC is re-induced in a subset of light-zone B-cells (centrocytes), allowing re-entry to the dark zone and maintenancy of the germinal center reaction
- Follicular lymphomas are tumors of germinal center B-cells (centrocytes and centroblasts) where the GC cells failt to undergo apoptosis, usually bc of chromosomal arrangement, t(14;18) that prevents normal switching off of BCL2
Post-GC memory B-cells circulate in the PB and account for at least some of the cells in follicular marginal zones of LN, spleen, and MALT
- express pan-B cell markers and surface IgM (with low levels of IgD) and are CD5- and CD10-
- Plasma cells made in the germinal center enter the PB and go to the BM, and have mostly IgG or IgA, lacking sIg and CD20 but expressing IRF4, CD79a, CD38 and CD138
- both memory B-cells and plasma cells have mutated IGV genes but do not continue to undergo mutation
- Post-GC B-cells maintain the ability to home into tissue in which they have undergone antigen stimulation so that B-cells made in the MALT tend to return there, and B-cells from LNs go to LNs or BM
- Marginal zone lymphomas of the MALT type, splenic type, and nodal type are Post-GC memory B-cells of marginal zone type that derive from and proliferate specifically in extranodal, splenic and nodal tissues
- Myeloma is due to BM-homing plasma cells
T-cells
T-cells come from a BM precursor that undergoes maturation and acquisition of function in the thymus
- antigen-specific T-cells mature in the thymic cortex
- Antigen-speicific T-cells mature in the cortex, and T-cells that recognize self-peptides are eliminated vis apoptosis
Cortical thymocytes have an immature T-cell phenotype and express TdT, CD1a, CD3, CD5 and CD7
- CD3 expressed in cytoplasm prior to complete T-cell receptor (TR) gene rearrangement and export to the cell membrane
- cortical thymocytes are originally double-neg CD4/8
- they are co-expressed in maturing thymocytes, and more mature T-cells only express CD4 or CD8
Medullary thymocytes have phenotype similar to mature T-cells
- 2 classes of mature T-cell in peripheral organs: alpha-beta T-cells and gamma-delta T-cells
- alpha beta and gamma delta chains are each made of a variable and a constant portion, and are assoc c the CD3 complex, which has gamma, delta, and epsilon chains
NK cells do not have a complete T-cell receptor complex; activated NK cells have epsilon and zeta chains of CD3 in the cytoplasm, and express CD2, CD7 and sometimes CD8, but not CD3
- they express CD16 and CD56, variably CD57, and have cytoplasmic cytotoxic granule proteins
- NK cells kill their targets with antibody-dependent cell-mediated cytotoxicity or another mechanism with killer activation receptors and inhibitory killer-cell immunoglobulin-like receptors
- NK cells do not rearrange TR genes, so antibodies to various killer-cell immunoglobulin-like receptor can be used for analysis of clonality in NK-cell neoplasms
- Toll-like receptors play role in cell-cell interactions and signaling, including recognition of infectious agents and initiating signaling through NF-kappaB
- are mostly in innate immune responses, but also have some role in the adaptive immune system
CD3+ / 56+ T-cell or NK-like T cells and gamma-delta T-cells are part of the innate immune system
- involved in mucosal and cutaneous immunity
- do not need APCs to initiate immune response (as in adaptive immune system)
Lymphomas of the innate immune system are usually extranodal in presentation, similar to the distribution of the functional components of this system
- many T-cell and NK-cell neoplasms are in younger patients
Gamma delta T-cells usually lack CD4, CD8, and CD5 expression; though a subpopulation is CD8+
- gamma delta T cells usually <5% of normal T-cells, and are usually restricted to the splenic red pulp, intestinal epithelium and other epithelial sites
- gd T-cellshave restricted range of antigen recognition and are the first line of defense against bacterial peptides, like heat shock proteins
T-cells of the adaptive immune system, which are heterogeneous and functionally complex, include naive, effector (regulatory and cytotoxic) and memory T-cells
- T-cell lymphomas of the adaptive immune system are seen in adults and nodal in origin, vs extrarnodal T-cell lymphomas of the innate immune system
CD4+ T cells are regulatory and act via cytokine production, based on cytokines are classified as T helper 1 (Th1) cells or T helper 2 (Th2) cells
- Th1 cells secrete IL2 and interferon gamma, but not IL4, IL5, IL6 or IL10; and provide help to other T-cells and macrophages
- Th2 cells provide help to B-cell with antibody production
- CD4+ cells can help to propagate or suppress an immune response
- T-BET and GATA3 have been found to be overexpressed in Th1 and Th2 lymphomas
T follicular helper (TFH) cells are a unique CD4+ T-cell subset in normal GCs; provide help to B cells in a GC reaction
- TFH cells express CD4, CD57, and CD279/PD1 and make the chemokine CXCL13 and its receptor CSCR5
- CXCL13 causes induction and proliferation of follicular dendritic cells and facilitates migration of B and T-cells expressing CXCR5 to the GC
- Increased CXCL13 is seen in angioimmunoblastic T-cell lymphoma (AITL)
-- AITL assoc c polyclonal hypergammaglobulinemia and expansion and proliferation of B cells and CD21+ follicular dendritic cells in a LN
Treg cells are another CD4+ T-cell subset which shut off and suppress immune responses; thought to prevent autoimmunity
- have high density of CD25 and transcription factor FOXP3, and also CD4
- T cell leukemia/lymphoma (ATLL) has been linked to Treg cells through CD25 and FOXP3 expression, and also explains the immunospreesion assoc c this dz
- The hypercalcemia in ATLL has been linked to osteoclast-activating activity


11 .01 The respective roles of various lymphocyte subpopulations in the immune system's two main arms: the innate and adaptive immune systems. In the innate immune system, which lacks specificity and memory, NK cells, NK-like T cells, and gamma delta T cells, along with other cells including granulocytes and macrophages, function as a first line of defence. These cells have cytotoxic granules (shown in red) containing perforin and granzymes. In the adaptive immune system, B cells and T cells recognize pathogens through specific receptors: immunoglobulins and the T-cell receptor complex, respectively. Antigen (Ag) presentation to T cells takes place via antigen-presenting cells (APCs) in the context of the appropriate major histocompatibility complex (MHC) class II molecule. Modified from Jaffe ES {1816}. [7]

Fig. 11.02 Normal B-cell differentiation and its relationship to major B-cell neoplasms. B-cell neoplasms correspond to various stages of normal B-cell maturation, although the normal cell counterparts are unknown in some instances. Precursor B cells, which mature in the bone marrow, may undergo apoptosis or develop into mature naive B cells, which, following exposure to antigen (AG) and blast transformation, may develop into short-lived plasma cells or enter the germinal center (GC), where somatic hypermutation and heavychain class switching occur. Centroblasts, the transformed cells of the GC, either undergo apoptosis or develop into centrocytes. Post-GC cells include both long-lived plasma cells and memory/marginal-zone 8 cells. Most 8 cells are activated within the GC, but T-cell-independent activation can take place outside the GC and probably also leads to memory-type 8 cells. Monocytoid 8 cells, many of which lack somatic hypermutation, are not illustrated. The red bars indicate IGH gene rearrangement and the blue bars IG light
chain gene rearrangement; the black insertions in red and blue bars indicate somatic hypermutation {3848).
CLL/SLL, chronic lymphocytic leukaemia/small lymphocytic lymphoma; D, surface lgD; DL8CL, diffuse large 8-cell lymphoma; FDC, follicular dendritic cell; M, surface lgM; MALT, mucosa-associated lymphoid tissue.[7]

11.03 B-cell differentiation. B cells go through various stages of differentiation as they mature from pro-B cell ls to plasma cells. The antigen-dependent phase of differentiation usually begins in the germinal center, where B cell ls encounter antigen. Red bar in nucleus indicates heavy chain gene rearrangement; blue bar indicates light chain rearrangement; black boxes connote somatic hypermutation. Cells colored in yellow have not encountered antigen, as opposed to antigen-dependent stages shown in violet. Modified from Swerdlow SH et al. {3848). BCR, B-cell receptor; D, surface lgD; M, surface lgM; SHM, somatic hypermutation. [7]

Fig. 11.06 Relative frequencies of B-cell lymphoma subtypes in adults. There are significant differences in the relative frequencies across geographical regions. However, diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma are the most common subtypes on a worldwide basis, but varying in some geographical regions and among some ethnic groups. Note that these estimates underestimate the incidence of chronic lymphocytic leukaemia I small lymphocytic lymphoma (CLL/SLL), because only patients presenting clinically with lymphoma were included. Data from the Non-Hodgkin's Lymphoma Classification Project {1}.
MCL, mantle cell lymphoma; PMLBCL, primary mediastinal large B-cell lymphoma [7]

11.04 T-cell differentiation. T cells mature in the thymus gland and then leave to occupy peripheral lymphoid tissues. T-cell receptor (TR) genes are shown schematically with a solid red bar indicating absence of rearrangement. The black boxes in the red bars reflect the rearrangements of the TR genes. The double red lines on the cell membrane represent the expressed T-cell receptor complex. Antigen dependent maturation leads to the different T-cell subsets, also illustrated in Fig. 11 .05. The phenotypes of several key T-cell subsets are illustrated:
T follicular helper (TFH), T regulatory (T-reg), T helper 1 (Th1), T helper 2 (Th2), and T helper 17 (Th17). Modified and updated from {3848). [7]

Normal B-cell maturation in the bone marrow. All plots show CD19-positive cells in a normal marrow specimen. Arrows follow changes in antigen expression with maturation from an early hematogone to a mature naive B cell. The earliest hematogones (stage 1) or B-cell precursors are shown in green and express strong CD10 with CD34 and CD38 with low CD45 without CD20. As B cells progress to stage 2 or intermediate-stage hematogones (shown in mauve), they drop CD34, and the level of CD38 increases slightly while CD10 and CD45 decrease and CD20 slowly increases. With progression to prenaive immature B cells or stage 3 hematogones (shown in orange), these cells continue to decrease expression of CD10 and acquire higher levels CD20 and CD45. With progression to mature B cells, CD20 and CD45 are acquired at high levels while CD10 and CD38 are lost. Note that B-cell markers CD24 and CD22 vary in intensity in a reciprocal way during this transition, with mature B cells expressing strong CD22 with low CD24 [18]

11.05 T-cell differentiation. T-cell neoplasms correspond to various stages of normal T-cell maturation. Mature T cells include alpha beta and gamma delta T cells, both of which mature in the thymus gland. Recently recognized T-cell subsets include the various types of CD4+ effector T cells, including T helper 1 (Th1), T helper 2 (Th2), T regulatory (Treg), T helper 17 (Th17), and T follicular helper (TFH) cells. Modified and updated from {3848). Ag, antigen; FDC, follicular dendritic cell. [7]

Fig. 11.07 Relative frequencies of mature T-cell lymphoma subtypes in an adult patient population. There are significant differences in the relative frequencies across geographical regions. However, peripheral T-cell lymphoma (PTCL), NOS, and angioimmunoblastic T-cell lymphoma (AITL) are two of the most common subtypes globally. Note that the category of enteropathy-type T-cell lymphoma (ETTL) used in this study is not equivalent to the category of enteropathy-associated T-cell lymphoma (EATL) as defined in this volume. The ETTL category was used largely as a generic category for most T-cell lymphomas involving the intestine, including EATL and more recently defined monomorphic epitheliotropic intestinal T-cell lymphomas. Data from the International T-Cell Lymphoma Project {4217).
ALCL, anaplastic large cell lymphoma; ATLL, adult T-cell leukaemia/lymphoma; E NK/T, extranodal NK/T-cell lymphoma [7]
Reactive Lymphocytosis
Main features of malignant lymphocytosis is that lymphocytes are atypical and monotonous [17]
- may not need to do flow cytometry, just if something doesn't seem right
Downey classification not really used in current practice, but may be useful to distinguish different types of lymphocyte morphology, which is more suggestive of a benign process
- Downey type I: less common, smaller cells, some nuclear indentation or a little lobation, may have cytoplassmic granules
- Downey type II: most common type of reactive lymphocyte, is a larger cell with round nucleus that can be a little irregular, with less condensed chromatin and abundant basophilic cytoplasm which wraps around RBCs. May see a slight nucleolus, but is not prominent.
- Downey type III: angry looking cells with coarse chromatin, these are frankly immunoblasts
LGLs can be seen in b9 and reactive conditions, but are usually <2.0 x 10^9 in reactive conditions
- assoc c BM transplantation, post-chemotherapy, and some viral infections
- neoplastic LGLs assoc c neutropenia, sometimes anemia
Transient stress lymphocytosis associated with trauma, burns, sickle cell crisis, myocardial infarction, and status epilepticus
- usually a mild lymphocytosis (4-10 x 10^9) and resolves within a day or two
Bordetella pertussis produces a toxin that prevents homing of lymphocytes from the blood back to lymphoid tissue
- mean lymphocyte count is 10 x 10^9, but can exceed 30 x 10^9
- classic morphology is cleaved or clefted lymphs
Persistent polyclonal B-cell lymphocytosis may persist for decades, and is associated with female smokers
- median lymphocyte count is 5.5 x 10^9, not very high
- polyclonal increase in IgM is another common findings, and 1/3 of cases have gain of isochromosome 3q
- morphologically, can see beautiful binucleated lymphocytes (different from pertussis)
Ddx for reactive lymphocytosis: mono, adenovirus, HHV6, rubella, roseala, mumps, chickenpox, cytomegaalovirus, parasitic infection (toxoplasmosis), drug reactions (anticonvulsants [phenytoin])

[17]





1 - 4
<
>
Cutaneous lymphoid hyperplasia in a 72-year-old man who presented with several purple-brown plaques on his cheeks and temples. A, The nodular infiltrate extends through the dermis with sparring of the overlying epidermis. B, It is characterized by scattered follicles with well-defined, reactive-appearing germinal centers, some of which have surrounding mantle zones. Many CD20-positive B cells are in the reactive follicles (C), and more numerous CD3-positive small T cells are present in the interfollicular areas (D). B-cell clonality studies (not shown) were negative for a clonal IGH or IGK gene rearrangement. [19]
Precursor Lymphoid Neoplasms
Overview
3 flavors:
- B-ALL/LBL NOS
- B-ALL/LBL c recurrent cytogenetic abnormalities
- T-ALL/LBL
Lymphoblastic lymphomas (LBL) vs Acute lymphoblastic leukemias (ALL) ??? If there is significant bone or BM involvement, ALL is used; if the tumor primarily involves an extramedullary site c little or no blood or BM involvement, LBL used
Both have undifferentiated blasts, and are neg for MPO and SBB; can be PAS+ in blocklike-coarse granular pattern
LBL is mostly T cell (in 4/5) and involves tissues and not BM or PB
- B-ALL is MC malig of kiddos (avg age 3 yo)
ALL usually B cell (4/5) and is diffusely in BM (>1/5), PB (>1/4) regardless of tissue involvement
- T-LBL usually anterior mediastinal mass; pts get hypercalcemia
Lymphoblastic lymphoma, the cytology not predictive of phenoty[e (left is B-cell, right is T-cell, or maybe vice versa? you would never know)

TdT is important in the diagnosis of lymphoblastic lymphomas, positive here in lymphoblasts around a germinal center (left) can also be used in cytology (right)

CD3 surface and cytoplasmic in T-ALL done by flow. CD99 is not specific, can also be seen in mature T-cell lymphomas

Differential diagnosis of Blastic neoplasms

B lymphoblastic leukemia / lymphoma (B-ALL/LBL), NOS
A malignancy of B-cell lymphoblasts
Comprise ~10% of lymphoblastic lymphomas
The rest are T lymphoblastic lymphomas
More common in children, most present as leukemia (10-20% as lymphoma)
Head and neck location is most common
If significant numbers of blasts in peripheral blood and/or bone marrow, it should be considered B-ALL
Micro: Usually diffuse pattern of involvement, with “single file” pattern of invasion into soft tissue
Cytology is typical for a “small round blue cell tumor”
High N:C ratio, condensed chromatin (though can look more finely granular and “blasty”, occasional nucleoli, frequent mitoses)
IHC: B cell markers (+ CD19/10/34/99, PAX5) nuclear TdT, HLA-DR, sometimes T cell markers (-/+ 13, 33, 117), 56
- negative sIg and CD20
- PAX-5 is most sensitive/specific in B-LBL; CD20 is variable; CD79a is usually positive, but can cross react with T-LBL; CD10 and TdT are positive in most cases
- if CD10 neg, suspect MLL gene anomaly
- up to 50% can have (+) myeloid markers (CD13/33)
- CD34 is variable
Genes: 6q, 9p, 12p abnormalities common
DDx: Hematogones disperse in CD34 stained marrow and in flow cytometry, while blasts tend to cluster
- by flow, hematogones have a wider range of expression (bright + CD10/20/34/81/TdT/sIg) while blasts are dimly positive
- B-lymphoblastic lymphoma of bone - can look similar to Ewing sarcoma, both are CD99+ and CD45 negative, but Precursr B-cell neoplasm CD45is TDT+, CD79a+, PAX5+ and CD43+
Px: low WBC count at dx is better, favorable if kiddos 2-10 yo, complete remission (on day 14 marrow) following induction, female, higher chromosome numbers (hyperdiploidy)
- hypodiploidy is unfavorable

B lymphoblastic leukemia / lymphoma, NOS

pre-B ALL
IHC of B-lymphoblastic lymphoma in LN, with TdT+ lymphoblasts surrounding negative follicle, they are positive for CD79a and PAX5, but negative for CD20.

B-lymphoblastic lymphoma of the bone vs Ewings sarcoma

B lymphoblastic leukemia / lymphoma (B-ALL/LBL) with recurrent genetic abnormalities
t(9;22)(q34;q11.2); BCR-ABL1
B-cell cancer with BCR on chr 22 fusing to ABL1 on chr 9, MC in adults
- no unique phenotype
IHC: (+) CD10/19/13/33, Tdt
- negative CD117
Genes: p190 kd fusion protein MC (7/10 BCR-ABL1 ALLs), versus the p210 kD protein made in most cases of CML, 2/2 less contribution of BCR gene to the fusion protein
Px: worst px of any lymphoblastic leukemia, might be slightly better in kiddos c chemo tx
t(v;11q23); MLL rearranged
MCC leukemia in kiddos <1 yo; the translocation even occurs in utero
- can occur in adults
- usually in the brain at dx; also presents c very high WBC count
Micro: nothing special
IHC: (+) CD19, 15, chondroitin sulfate proteo-glycan neural-glial antigen 2 (NG2)
- negative CD10, 24
Genes: MLL on 11q23 can fuse with a bunch of different spots, but usually comes from cr 4
Px: MLL-AF4 has a poor px
t(12;21)(p13;q22) TEL-AML1 (ETV6-RUNXT1)
usually in kiddos
IHC: (+) CD19/10/ 13 (weak) /33 (weak),
- negative: CD9
Px: Good
Hyperdiploid
has >50 chromosomes; extra copies usually of 21, 4, 14, X
- in kiddos
IHC: (+) CD19/10/34, TdT
Px: Good
Hypodiploid
has < 46 chromosomes; in kiddos and adults
IHC: (+) CD19/10/34, TdT
Px: Poor
t(5;14)(q31;q32) IL3-IGH
Rare; in kiddos; has eosinophilia
- chromosomal rearrangement results in the overexpression of IL3 under the promoter of IGH and the excess of cytokine results in eosinophilia
- the association is so strong that a disease relapse is often heralded by increasing eosinophil counts [21]
IHC: (+) CD19/10/34, TdT
Px: ok
t(1;19)(q23;p13.3) E2A-PBX1 (TCF3-PBX1)
in kiddos
IHC: (+) CD19/10
- negative CD34
Px: Poor
B-lymphoblastic leukemia/lymphoma, BCR-ABL1-like
Practical approach to identification
– Selecting an optimized screening approach and defining the appropriate population to screen is a challenge
• High index of suspicion
• If available, CRLF2 expression by flow
• Advanced applications
– Specific phenotype using small set of genes, FISH probes or molecular methodologies (LDA, RNA seq)
Incidence: relatively common (~20%)
– Higher in children with high risk ALL, adolescents and adults
– Down syndrome, high rate of CRLF2 translocations
– IGH/CRLF2 translocation common in Hispanic and Native American populations
• Clinical, morphologic and immunophenotypic features similar to other subtypes of B-LL
– Often present with higher WBC counts
– CRLF2 expression by flow distinctive
• Lacks the BCR-ABL1 fusion/Philadelphia chromosome, BUT
• Shows a gene expression profile that is very similar to B-LL with BCR-ABL1
• Because of the clinical importance of identifying these cases, added as a distinct WHO entity – 2017
• Overall poor prognosis
• High risk of poor response to therapy and relapse
Genes: Although heterogeneous, converge on activation of kinase signaling pathways
– JAK/STAT pathway
– ABL-class genes
– Increased signaling due to IKZF1, FGFR1, RAS
• Some of these translocations may be detected by routine cytogenetic analysis but others are cryptic
JAK/STAT pathway activation
– Cytokine receptor-like factor 2 (CRLF2)
• 50% of cases of Ph-like cases
• Majority due to P2RY8-CRLF2 and IGH-CRLF2
• Typically show high CRLF2 levels by flow cytometry
– JAK - Activating point mutations (30-50% of CRLF2-rearranged cases)
• JAK rearrangements (5-7%)
– Erythropoietin receptor (EPOR)
• 1-5% of cases
• Protein overexpression
ABL class abnormalities
– Typically results in fusion genes as a consequence of translocations/similar rearrangement
– e.g. ABL1, ABL2, CSF1R, PDGFRA, PDGFRB
– Create constitutive activation of tyrosine kinase signaling proteins
• Other
– IKZF1, PAX5, FGFR1, mutations, deletions, etc.
– More than 60 described thus far
– Key: results in GEP similar to BCR-ABL1
Treatment implications
• CRLF2 alterations: could be a candidate for JAK2 inhibitors given the activation of the JAK/STAT pathway
• Tyrosine kinase abnormalities: amenable to/potentially responsive to TKIs
• JAK mutations/translocations may be candidate for JAK inhibitors

B-lymphoblastic leukemia/lymphoma with iAMP21
• Recent addition to WHO 2017 due to its propensity to relapse if treated as standard risk ALL
• ~2% of childhood ALL
• Often accompanied by low WBC count
• No unique morphologic features
Typically recognized (by default) using the FISH probes to detect the cytogenetically cryptic t(12;21); ETV6-RUNX1
• Defined as amplification of a portion of chromosome 21, often >5 copies of the RUNX1 gene region
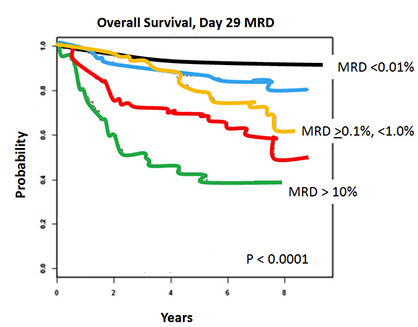
Survival curves
• Patients with iAMP21 have inferior outcomes with the less intensive chemotherapy regimens (eg, SR) top graph
• In contrast, the outcome of high risk iAMP21 patients was not significantly different from those lacking iAMP21

iAMP survival curve

T lymphoblastic leukemia/lymphoma (T-ALL/LBL)
Cancer of lymphoblasts in the T-cell lineage,
- M>F, ~15% of ALL's in kiddos and almost 1/2 of childhood lymphomas; also 1/4 adult ALL's are T-cell
- usually in anterior mediastinum c involvement of other organs
- up to 9/10 of all lymphoblastic lymphomas (rest [10%] are of B-cell lineage - compare c ALL...)
- call it just a lymphoma if there is no leukemic component
For some treatment regimens, >25% blasts in BM to define leukemia
Micro: medium sized c scant cytoplasm, inconspicuous nucleoli
- PB has cells c delicate chromatin convoluted nuclear membranes, freq mits, sometimes cytoplasmic vacuoles
- Lymphoma (T-LBL) can look like starry sky, c lots of pericapsular and soft tissue infiltrates
IHC: (+) cytoplasmic CD3, CD2/ 99 (var) / 7, occasional NK / myeloid (CD13/33) markers, TdT nuclear, variable CD3 / 5 / 34 (var) / 10 / 1a / 45
- negative CD19/20, HLA-DR
- either CD4/8 double + or neg
Genes: TCR rearrangements, 1/5 c simultaneous IGH@ rearrangements
- up to 3/4 c abnormal karytypes, such as 14q11.2, 7q35, and 7p14-15
- NOTCH1 mutations (1/2) assoc c worse px just in adults (not kiddos)
DDx: thymoma has epithelial markers such as EMA, and has marked dispersal of plots of CD4 vs CD8 (marked coexpression) and CD45 vs CD3 plots
- clustering is more tight in T-LBL
Px: Agressive, c 1/4 5 yrs survival; though ~1/2 can be cured c chemo
- worse than B-ALL's
- px depends on age, stage and LDH levels

T-LBL

Early T-cell precursor lymphoblastic leukemia (ETP-ALL)
New entity: Single subtype of T-ALL/LBL recognized specifically by the WHO due to its unique immunophenotyped and relative paucity of T-cell antigen expression
• All other cases of T-ALL diagnosed as such
• ~10% of childhood ALL cases (1-2% of all ALL)
• No distinct cytologic features
IHC: CD7 and cCD3 positive, neg CD8 and CD1a, partial/weak CD5 (if expressed) AND express one or more myeloid/stem cell markers (CD34, CD117, CD13, CD33, CD11b, CD65, HLA-DR)
• Myeloperoxidase is negative (by definition)
Genes: Originally recognized in 2009 from gene expression profiling data
– represents a subset of T-ALL sharing transcriptional and immunophenotypic similarities with early T-cell precursors (Coustan-Smith et al., 2009)
– Gene expression profile is different from T-ALL at later stages of maturation
• Myeloid/stem cell profile resemblance
– Overexpressed genes (eg, CD34, CD117, CEBPA)
– More frequent mutations in FLT3, DNMT3A, IDH1, IDH2
Px:
Original early studies showed a worse prognosis compared with typical T-ALL (2009 St. Jude)
• A more recent study shows differently (2014 COG) (Wood, BL, Winter SS, et al Blood; 124:1 (abstract))
• More recent study again shows unfavorable prognosis (Jain N, et al. Blood 2016. PMID 26747249)



CD3 neg (dim poositive may be artifact)
CD3 neg, some cells are weakly positive
Cytoplasmic CD3 clearly positive
3 - 3
<
>
Indolent T-lymphoblastic Proliferation (iT-LBP)
Often associated with other pathologic conditions, including AITL, Castleman disease, follicular dendritic cell sarcoma, and associated with paraneoplastic autoimmune multiorgan syndrome, and carcinomas such as hepatocellular carcinoma or acinic cell carcinoma
- typical anatomic sites include the upper aerodigestive tract, cervical, supraclavicular, abdominal, or retroperitoneal locations
Micro: the blasts of iT-LBP usually form sheets or dense clusters within the interfollicular / paracortical areas of lymph node, with preservation of the general follicular lymphoid architecture
- if associated with carcinoma, the blasts intersperse or cluster between carcinoma cells without lymphoepithelial lesions
IHC: has the immunophenotype of immature thymocytes, (+) TdT, CD3, CD4, CD8, and variable but generally positive CD10, CD99, and CD1a
- variable CD2, CD5, and CD7
- neg CD34
DDx: T-LBL usually effaces the entire node or shows partial infiltration obliterating residual follicules
Tx: not required; does not behave like an aggressive lymphoma; although need to treat associated diseases
Minimal residual disease in ALL
• Key prognostic indicator
• Must distinguish normal from abnormal
• Can be done by flow or molecular methods
– Flow tends to be faster
– Molecular can be more sensitive
• Key considerations: specimen quality, targeted therapies and potential effect on FC detection
NK-lymphoblastic leukemia / lymphoma
-
Mature B-cell neoplasms


Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL)
Monomorphic, mono-/oligoclonoal small round B-lymphocytes in lymph tissue (spleen, LNs), BM, PB which are CD5/23 (+)
- MCC leukemia in adults in Western countries (very low in Eastern countries [= genetically inherited?]); 2:1 M:F ratio; avg age 65 years
- (+) FamHx at least doubles risk of CLL (strongest genetic influence of all B-cell cancers; familial clustering in 1/20; 1st degree relative = 5x baseline risk)
- sx= adenopathy, S-megaly, PB/BM involvement
- autoimmune dz common, (+) DAT in 1/3
- immunodef common; hypogammaglobulinemia in up to 1/2
- sometimes see M protein
Defined as Monoclonal B-cell count > 5 x 109/L in peripheral blood
- Characteristic immunophenotype
- The term small lymphocytic lymphoma is used when there is diagnostic extramedullary involvement and the PB count is < 5 x 109/L
Monoclonal B-cell count?
% Lymphs of clonal population x absolute lymph count
ALC = 16.2 (46%) = 7.5
MBCL = 7.5 (0.71) = 5.2
Really 2 diseases
1) Post-germinal center - has hypermutated IgVH
- CD38 and Zap70 neg
- Px: Favorable
2) Pre-GC - non-mutated IgVH
- (+) CD38 and Zap70
- Px: Adverse
Lymphocytes must be elevated for 3 months to dx; unless pt has sx or other cytopenias
"SLL" used for CLL localized to tissues (without leukemia), and requires lymphadenopathy and no cytopenias from BM infiltration
Micro: LN c diffuse nodal involvement showing pseudofollicular architecture (no true mantle zone) c large pale prolif centers c prolymphocytes (>11%; sm to med size cells c clumped chromatin and small nucleoli), paraimmunoblasts (larger, c round to oval nuclei, central red nucleoli)
- 11-55% prolymphs = CLL/PLL
- can get rid of most smudge cells c heparin
CLL/SLL can have a perifollicular and/or follicular growth pattern
Smudge cells seen in PB with small lymphs c clumped chromatin and round nuclei
- lots of prolymphocytes in PB = poor px (>55% prolymphs = B-cell prolymphocytic leukemia (B-PLL)
BM typicaly has at least the finding of >30% lymphs; not well established criteria
- can see prolif of small, mature lymphs c multiple prolif centers / pseudofollicles
IHC
(+) IgM/D2, CD5 (>3/4)/11c/19/20 (dim) / 22/23/43/79a, Bcl-2, PAX5
- neg: CD10/79b/81, FMC7 (dim to neg), cyclin D1 (Bcl1)
-- proliferation centers can be cyclin D1+ in up to 30% of CLL/SLL
- CD23+/FMC7- can differentiate CLL/SLL from MCL in most but not all cases
- atypical profiles have CD5/23 (-); CD11c/79a/FMC7
Should ALWAYS do Cyclin D1 on new cases of CLL to make sure it is not mantle cell lymphoma
- having some Cyclin D1 positivity in the proliferation centers can be seen in up to 30% of cases , especially in the subset of CLL/SLL with CONFLUENT proliferations centers (tend to behave more aggressively)
- some mantle cell lymphomas can have small round nuclei
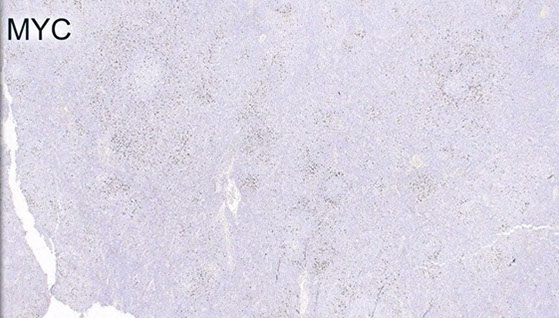
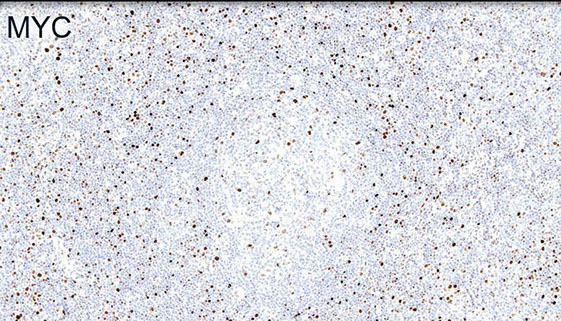
MYC is often increased usually in proliferation centers (sometimes with inc Ki67)
New IHC markers in CLL:
LEF1 - a highly specific martker for CLL/SLL (+ in ~100%, but also + in some larger lymphomas (DLBCL and HG-FL) and is positive in T-cells
CD200
Useful diagnostic marker
Bright in CLL, dim/- in mantle cell lymphoma
CD49d
Useful prognostic marker (better than CD38)
>30% of clonal B-cells+ = CD49d+
CD49d+ 10 yr OS probability 62% vs 84% for CD49d- (Bulian et al. J Clin Oncol . 2014;32:897-904)
Flow: small FMC7 neg B-cells, CD20 dim, light chain dim
Genes: rearranged IG genes unmutated in 1/2, and mutated in 1/2 of cases; are clinically different
- tyrosine kinase ZAP-70 or CD38 expression assoc c unmutated CLL IGHV gene (has not gone through the GC), and thus an aggressive clinical course and poor px
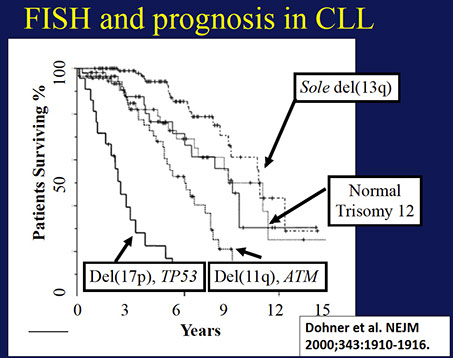
80% have FISH abnormality (50% have del13q14.3, 20% trisomy 12)
- BM involvement has poor px
- trisomy 12 has intermed px
Genes: MC abnormality is normal (1/2), then trisomy 12 (1/3, atypical cells, adverse px), then del13q14 (~1/4, has good px)
- large number of mutations that occur with low freq
- FISH may have del 13q14 (>1/2), also tri 12, del(11q), del(14q), del(17p); 1/5 normal by FISH
-- del 13 and trisomy 12 have good px, del 11q and 17p have a poor px
Px: uses Rai/Binet staging system
- Pts c mutated CLL (IgHV [see below]) have better px than unmutated CLL
- (+) ZAP-70 and CD38 assoc c poor px; VH3-21 also poor px; 11q deletion is poor px
- CLL cells have uniform Ig heavy chain variable (IgHV) genes, which most likely came from single precursor cell prolif.
- px affected by high stage, B-sx, diffuse BM involvement, doubling time <1 yr in PB, high initial lymph count (>30k), unmutated Ig HC variable region (IgVH) [ CLL c unmutated IgVH resembles pregerminal center B-cells, is likely to progress, poor px, can be tx'd, have ZAP-70/CD38 expression]
- Proliferation centers that are large / confluent and have a high proliferation fraction are a significant and independent adverse prognostic indicator
Highly Aggressive CLL
Not a specific category of CLL but mentioned in the 2017 revised 4th ed. of WHO classification.
•Aggressive histologic features:
1.Prominent proliferation centers broader than 20x field or becoming confluent.
2.Ki-67 proliferation index >40%, or >2.4 mitoses in proliferation centers.
•More commonly associated with higher LDH levels and ZAP expression than non-accelerated cases.
•Outcome intermediate between CLL and Richter syndrome (DLBCL).
•Not considered large cell transformation.
Select molecular mutations in CLL and prognostic effect
TP53 (4-10%)
- May occur alone in 25–30% of cases; more frequent in combination with a loss of the other TP53 allele (deletion)
- Adverse prognosis (similar to deletion)
NOTCH1 (10%): Associated with poor outcome
SF3B1 (4-9%): Associated with more aggressive disease and poorer clinical outcome
BIRC3 (4%): poor outcome
Rosati E, Baldoni S, De Falco F, et al. NOTCH1 Aberrations in Chronic Lymphocytic Leukemia. Front Oncol. 2018;8:229. ; Buccheri V, Barreto WG, Fogliatto LM, Capra M, Marchiani M, Rocha V. Prognostic and therapeutic stratification in CLL: focus on 17p deletion and p53 mutation. Ann Hematol. 2018;97(12):2269–2278; Wan, Y., & Wu, C. J. (2013). SF3B1 mutations in chronic lymphocytic leukemia. Blood, 121(23), 4627–4634; Rossi D, Fangazio M, Rasi S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood. 2012;119(12):2854–2862.
Karyotype in CLL
Complex karyotype associated with worse survival
How to define (>3, >5?)
Worse prognosis
However, up until relatively recently cytogenetic analysis was never widely incorporated into the routine diagnostic algorithm of CLL
- mainly due to technical considerations
- relative difficulty in obtaining sufficient metaphases of the CLL clone; with subsequent low detection rate of chromosome abnormalities
IGH mutations
Frequency approximately 8%
Variety of partners:
- BCL2/t(14;18)(q32;q21)
- BCL3/t(14;19)(q32;q13)
- MYC/t(8;14)(q24;q32)
- BCL11A/t(2;14)(p16;q32)
Differential outcomes of BCL2 and BCL3 in CLL
Fang et al: OS of IGH‐BCL3 group was significantly shorter compared to the Dohner high risk non‐IGH group (P = 0.01)
Up to ~10% of pts develop DLBCL (Richter transformation), and <1% evolve to Hodgkin lymphoma
- transformation to DLBCL has <1 yr survival rate
- MC transformation is to PLL or CLL/PLL
- Clonally related (most cases): IgHV unmutated; median survival < 1 year.
•Clonally unrelated: prognosis identical to de novo DLBCL; may arise secondary to immunosuppression/immunodeficient; oftern EBV+.
•Associated with TP53 abnormalities, NOTCH mutations, CDKN2A deletions, and MYC translocations.
Transformation to Hodgkin is uncommon, <1%, Requires classic Reed-Sternberg cells in appropriate cellular background.
Most common subtype = mixed cellularity

CLL, proliferation centers (pseudofollicles), usually MUM1+
from LASOP lecture, 1/18/2020

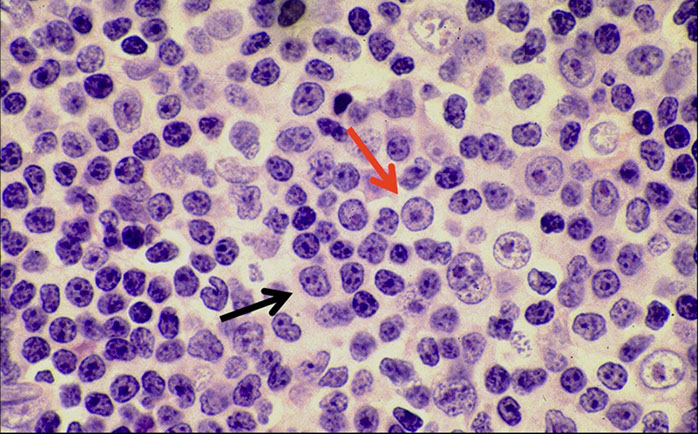
CLL pseudofollicle (proliferation center). Little lymphocytes with small cytoplasm on the left. On the right, paraimmunoblasts are larger, with a little more cytoplasm, with prolymphocytes (black arrow)showing dispersed chromatin, and the red arrow to paraimmunoblasts with prominent central nucleolus (called "paraimmunoblasts" because typically smaller than B-type immunoblasts).


LASOP lecture Dr Reichard, 1/18/2020


"Accelerated" CLL, with Ki67 >40% in proliferation centers can have px bwt CLL and DLBCL


The "MZ look" found in some cases of CLL/SLL, looks like proliferation centers surround germinal centers

CLL/SLL can show nuclear irregularity
Interest in BTK gene and PI3K mutations for therapy [9]

CLL IHC. CD20 (upper left), CD3 strangely highlighting some of the GCs (upper right), CD5 (lower left) shows staining of at least a few of the B-cells, and CD43 (lower right) is positive on many of the B-cells and shows a small ring of negative cells . around the proliferation center in mantle zone area (black arrow)


Ki67 Showing a small ring around proliferation centers in mantle zone area (same case as IHC to the left)
CLL/SLL IHC. Follicular dendritic cells and neoplastic B cells are positive for CD23 (upper right), the FDCs and B-cells are strongly positive for IgM (upper right), and are Kappa-restiricted (bottom images)


CD279/PD1 T-cells strongly + in Germinal Centers (GC)
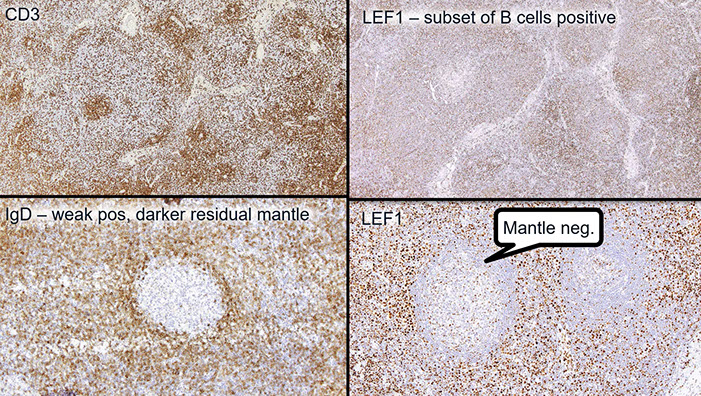
CLL IHC. CD3 positive (upper left), LEF1 staining a subset of B-cells (upper right), IgD weakly positive in the tumor cells and more strongly (darker) positive in the residual mantle zone (bottom left), with LEF1 negative in the mantle zone (bottom right)
CLL IHC. In this case, MUM1 with weaker staining on some B-cells and was strongly positive in some areas(upper image), Ki67 showing more positivity in the germinal center-like areas, less positivity in the mantle-cell areas, and then moderate proliferation in the interfollicular areas (bottom image)

MYC more strongly positive in proliferation center-like areas
Monoclonal B-cell Lymphocytosis (MBL)
Monoclonal B-cell population in PB up to 5x10^9/L
- can have phenotype of CLL, atypical CLL, or non-CLL (CD5-)
- MBL is very rare under 40 years - seen in 75% of patients older than 90 years
- is a precursor for CLL/SLL
- may have BM involvement without a maximum level of involvement, extent of marrow involvement not a criterion in the differentiation between CLL/SLL and MBL [9]
MUST distinguish between Low and High-count MBL
- "low count" MBL (<0.5x10^9/L) is different from "high count" MBL bc the low-count type has not been shown to progress to CLL/SLL
-- high-count MBL requires routine f/u
Extent of bone marrow involvement is not a criterion in the ddx bet CLL/SLL and MBL!
- if you don't know that the patient has >5 x 10^9 PB CLL cells or other evidence of lymphoma, need to make a wimpy diagnosis! [9, Swerdlow lecture]
- Asymptomatic expansion of clonal B-cells
- Estimated to be 100X more common than CLL
- Precursor state to CLL; about 1-2% progression rate per year to CLL
- Prevalence of MBL varies based on the population studied and sensitivity of the flow cytometric analysis - up to 20.9% for CLL –type in 1 study
Must know CBC data and clinical information to interpret flow results
- Must distinguish from small lymphocytic lymphoma with minimal PB involvement and treated CLL
MBL Subclassification
CLL-like phenotype (>75% of cases)
– CD5+, dim CD20, dim sIg
Atypical-CLL phenotype
- CD5+ but bright CD20, bright sIg or CD23(-)
Exclude mantle cell lymphoma by FISH
Non-CLL phenotype
– CD5(-), bright CD20 and bright sIg
- some may be transient and self-limited, additional cytogenetic studies needed to rule out a specific lymphoid neoplasm,
- may be related to splenic marginal zone lymphoma
- some have aberrance 7q
MBL diagnostic criteria
Monoclonal B-cell count <5 x 109 cells/L in the peripheral blood
No other features of a lymphoproliferative disorder or autoimmune disease
– Normal physical exam (no lymphadenopathy or organomegaly)
– Non-Hodgkin lymphoma excluded
– Absence of disdisease-related symptoms and cytopenias
Marti GE, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br J Haematol 2005; 130: 325–332.
Hallek M, et al. Guidelines for
Lymph node infiltration by CLL-type cells without Proliferation Centers (PC) in the absence of lymphadenopathy >1.5 cm on CT scan who otherwise have MBL "may represent a nodal equivalent of MBL rather than SLL" [9, also from blue book], though this is based on a single retrospective study with relatively short follow up

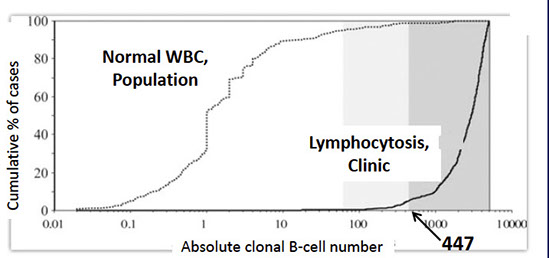
Marked difference in the clonal B-cell count in CLL-type MBL vs population studies
• Median 1 clonal B-cell per μL in population studies compared to median 2,939 clonal B-cells per μL in clinical hematology series
• 95% of CLL-type MBL cases have more than 447 clonal B-cells per μL highlighted by the dark grey background
Rawstron AC, Shanafelt T, Lanasa MC, et al. Different biology and clinical outcome according to the absolute numbers of clonal B-cells in monoclonal B-cell lymphocytosis (MBL). Cytometry B Clin Cytom. 2010;78 Suppl 1:S19–S23.
From Dr. Reichard LASOP lecture, 1/18/2020
Survival curves for patients with MBL involving a lymph node, showing that larger lymph node size (>1.5 cm) and presence of proliferation centers (PC) are associated with worse outcomes [9]


B-cell prolymphocytic leukemia (PLL)
As name implies, is a proliferation of prolymphocytic B-cells in the PB/BM/spleen
- presents c very high WBC count (>100k), c B sx, cytopenias and S-megaly
- must have >55% prolymphocytes in PB
-- must exclude transformed CLL, CLL c inc prolymphocytes, and other malignancies that may look similar but have t(11;14)(q13;q32)
Prolymphocytes are 2x the size of normal lymphs c prominent nucleolus and a moderate amt of lightly basophilic cytoplasm
In spleen see expanded white pulp that infiltrates into the red pulp
- no pseudofollicles (prolif centers) are present
Difficult to r/o other cancers by morphology, need cytogenetics
Prolymphocyte IHC profile:
IgM (+)/ IgD (-), (+) B-cell markers (CD19/20/22/79a,b, FMC7)
- CD5/23 seen in ~20% of cases
- ZAP70 and CD38 seen in ~50% cases
Ig genes and unmutated heavy chain genes rearranged in ~50% cases
- all B-PLL cases are part of either the VH3 or VH4 gene family classes
- Must exclude cases c t(11;14)(q13;q32)
Del(17p) seen in ~50% of cases and is assoc c TP53 mutations
Poor response to tx; no good, clear-cut prognostic indicators
- live from 2 to 4 yrs on avg
Splenectomy may help improve sx
B-PLL, peripheral blood
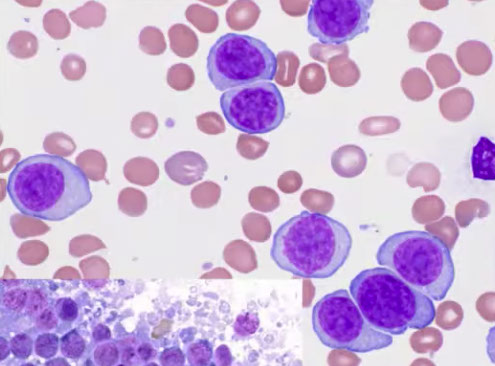
B-PLL, touch imprint


Usually HCL is CD10 negative
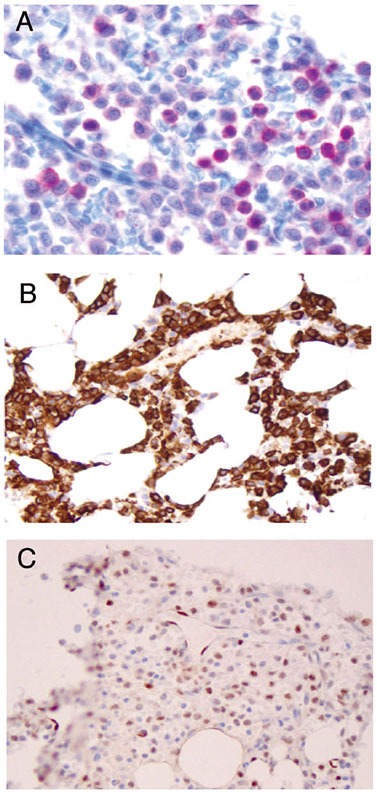
Expression of annexin A1, DBA-44, and BCL1 by hairy cell
leukemia (HCL). This HCL is the same case shown in Figure 4.A through C, The atypical lymphoid cells are positive for annexin A1 (partial) (A), DBA- 44 (B), and BCL1 (partial) (C) (original magnification3400 [A]; original magnification x200 [B]; original magnification x200 [C]).
- these markers


Hairy cell leukemia (HCL)
Indolent cancer of B-cells with "hairy" cytoplasmic projections, oval nuclei and lots of cytoplasm
Tumor cells found mainly in BM and spleen, may involve other organs
HSmegaly, recurrent infx, circulating neoplastic cells and monocytopenia characteristic
- can present c new neutropenia, monocytopenia or aplastic anemia
- 4M>1F, whites>blacks
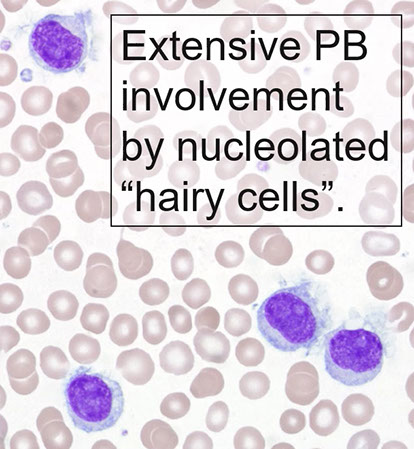
PB: large lymph (double size of normal lymph) c round or reniform nuclei c smooth contours, ground glass chromatin c indistinct to absen nucleoli
can have circumferential hairy projections caused by frayed cytoplasmic edge
BM: widely-spaced infiltrates of hairy cells; no mits; lots of fibrosis (usually a dry tap) and hypocellular marrow
Tissue shows "fried egg" morphology, reticulin fibrosis, blood lakes, mast cells frequent
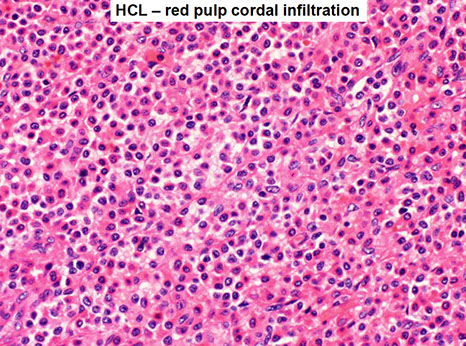
- Spleen white pulp is atrophic/absent c red pulp cords infiltrated c tumors that blocks circulation
Cells look hairy b/c of overexpression of Rho family of small GTPases and Gas7 molecule up-regulation (a growth-arrest molecule)
Patchy distribution of cells due to activiation of motility receptor which binds to things it shouldn't
HCL makes fibronectin and has bFGF and TGFB1 secretion by hairy cells which may cause reticulin fibrosis of BM
CCR7 and CXCR5 not expressed, and integrin and matrix-metalloproteinases overexpressed which explain the absence of hairy cells in LNs and presence in spleen/BM respectively
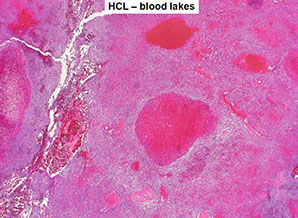
In spleen, see BLOOD LAKES
IHC: TRAP is a technically difficult stain to perform
(+) CD20/22/11c/103/25/123, T-bet, annexin-1 (ANXA1), DBA.44, FMC-7, Cyclin D1 (weak), HBME-1, cyclin D1 (weak, and does not harbor the genetic mutation)
(-) CD5 / 10 (+ in 10-20%)/ 23 / 43
- can rarely be CD10+ (need >30%+ cells threshold to call +)
Flow: CD20 bright, CD22 bright, CD103+, CD11c+, CD25+, very few monos in PB/BM
- ANXA1 may be most specific marker (though must be careful bc is expressed by myeloid and T-cells; which is why ANXA1 cannot monitor MRD)
Should have the ABCs of HCL to distinguish from mimickers: Annexin A, BRAF and Cyclin D1
- CD103 and CD11c by itself not specific enough
~90% have VH genes c hypersomatic mutations (from post-germinal center maturation)
Multiple clonally related Ig isotypes commonly expressed, indicating maturation arrest during isotype switching
Nearly all cases of HCL have somatic BRAF V600E activating mutation in the serine / threonine kinase BRAF leading to constitutive activation of RAF-MEK-ERK signaling pathway which prolongs cell survival
- BRAF V600E is not present in HCL variant, although a high percentage have IGHV4-34 Ig rearrangements, with mutations in MAP2k1 whick encodes MEK1, leading some to postulate that HCL variant has a different pathogenesis from HCL
Sensitive to alpha interferon and pentostatin/cladribine (purine analogs), often achieving complete remission
- may (uncommonly) get complete remission from splenectomy
- adding rituximab may help in relapse
EM: ribosime lamellar complexes
Px: >90% 10-year survival
- survivors have inc % of secondary neoplasms
HCL gross - Diffuse red pulp enlargement, blood lakes

HCL blood lakes
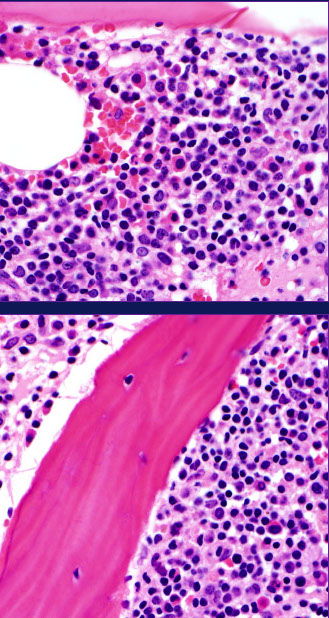
HCL IHC

Splenic B-cell lymphoma/leukemia, unclassifiable
Small B-cell clonal proliferations of spleen that don't fit another category
- mostly are splenic diffuse red pulp small B-cell lymphoma and hairy cell variant
-- both are DBA.44 and IgG (+), IgD (-)
Splenc diffuse red pulp small B-cell lymphoma (SDRPL)
May look similar to Hairy-cell leukemia variant, with blood lakes in spleen and sinusoidal pattern of BM involvement; however, usually does not have a high degree of lymphocytosis, like that seen in HCL-v
Micro: diffusely involves red pulp, sparing white pulp
IHC: (+) CD20, CD11c, DBA.44, CD103 (15-40%)
- negative Annexin A1, CD23, CD5 (up to 30%), CD123 (positive in 16%)
Genes: not specific, BRAF V600E usually negative
- mutations in NOTCH1, MAP2K1, TP53 associated with aggressive behavior
DDx: SMZL, HCL, HCL-v
Tx: not curable, though patients usually do well with splenectomy and anti-CD20 treatment
SDRPL IHC

Hairy cell leukemia, variant (HCL-v)
Resembles HCL but should not be considered a type of HCL
- BRAF V600E mutations seen in almost all HCL but not in HCL-v, and Annexin A1 is negative
- has different cytologic and IHC
- comprise ~10% of HCL cases; is not biologically related to HCL
- cells in PB looks like mix of HCL and prolymphocytic leukemia
- not as much fibrosis or infiltration in BM, though spleen still diffusely involved
IHC: May lack characteristic stains of HCL (CD25, ANXA1, TRAP, CD123) but are still often CD103 (+)
Genes: high prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing HCL (both aggressive) [Nature Genetics, 2014, 46:8]
Px: Have indolent course and long survival, even though doesn't respond to same tx as HCL
- rituximab and anti-CD22 may be highly effective
- splenectomy still advised
HCL-v
Lymphoplasmacytic Lymphoma (LPL)
Cancer c small B-cells, plasmacytoid lymphs, and plasma cells often in BM that does not fulfill the criteria for any other small B cell lymphoid neoplasm with plasmacytic differentiation
Many small B-cell lymphomas can have plasmacytic differentiation -- LPL is a diagnosis made after excluding other lymphomas with plasmacytic differentiation:
- Marginal zone lymphomas (nodal, extranodal/MALT, splenic)
- CLL/SLL
- Follicular lymphomas
- Mantle cell lymphomas (very rare)
Difference between this and other neoplasms not always so clear, may need to just say "Small B-cell lymphoma c plasmacytic differentiation" and give ddx
- Plasmacytic neoplasms can co-exist with lymphoid neoplasms -- not "lymphoplasmacytic" and some PCM can appear quite lymphoid
- there are HCV-associated clonal lymphoplasmacytic proliferations that may not behave like a neoplasm (but cases of LPL can be associated with HCV)
WHO 2017 updates:
In 2008 WHO, no recurring chromosomal or oncogene abnormalities are recognized
•In 2017 WHO,
– MYD88 L265P mutation in >90% of LPL or Waldenström macroglobulinemia
– CXCR4 mutations are found in about 30%
• MYD88 mutations are not however entirely specific for LPL and can be seen in a small % of other low-grade B-cell disorders
Frequently have IgM paraprotein and Waldenstrom's macroglobulinemia (WM, defined as LPL in BM c IgM monoclonal); 30% of pts have hyperviscosity
- up to 20% of pts c WM get it genetically (usually in younger pts, BM looks worse)
- IgM can deposit in skin and GI
Hep C assoc c LPL and type II cryoglobulinemia; these cases respond to antiviral tx
Classically (when assoc c WM), LN shows sinuses dilated c PAS(+) material
- also see Dutcher bodies (PAS[+] pseudonuclear inclusions), inc mast cells and hemosiderin
-proliferation centers (as in CLL/SLL) absent, paler well-defined marginal zone also absent
Micro: spectrum of small lymphs to plasma cells
- can see Dutcher bodies
- LN architecture can be preserved (if in interfollicular spaces and sinuses) or can be effaced
- can see PAS+ material in sinuses
IHC:
(+) IgM (patchy, sometimes IgG or IgA), CD19 / 20 / 22 / 25 / 38 / 138 / 79a, Bcl-2 (var), sIG (K or L), PAX5, MUM1
(-) CD5/ 10 / 23 / 43 (1/5+) / 103 / 56, Bcl-6, EBV/HIV
Immunophenotype [9, lecture 2]:
- Light-chain restricted lymphocytes (flow cytometry, PSIP only in some labs) and light chain restricted plasma cells (PSIP)
- Usually IgM+, but sometimes IgG+ and rarely IgA+. IgD is not found in most cases
- CD5- (Mayo Clinic study 43%+), CD10- (some exceptions....esp at slide workshops), usually CD23- (but some find a moderate # of + cases)
- often CD25+, CD138+
- Plasma cells are CD138+
Genes: MYD88 mutation is NOT specific (lots of other B-cell neoplasms have it), do not diagnose LPL solely on a MYD88 mutation
- other B-cell lymphoma-associated translocations NOT found (eg, CCND1, MALT1, BCL10, BCL2 [possible rare exceptions])
- infrequent trisomies 3, 12, 18, but trisomy 4 in 20%
- 6q21 deletions occur in up to 63% of BM-based LPL (not specific)
- gene profiling studies suggest a homogeneous gene expression profile independent of 6q deletions and more like CLL or normal B0cells than myeloma
- MYD88 L265P mutation positive in most (90%) cases of LPL , (including 3/3 non-IgM secreting -- Ig-2 and IgA-1but not specific (see image) [6]
- mutation is also seen in a significant proportion of IgM but not IgG or IgA MGUS cases, different types of DLBCL, but not in plasma cell myeloma of any type
- may not be possible by morphology and IHC to separate from MZBCL, in which case the +MYD88 is useful to dx LPL
Px:
- Indolent course c 5-10 year survival
- Poor px: older pts, severe cytopenias, high B2-microglobulin, possibly increased immunoblasts / transformed cells, del(6q)
A more aggressive variant is gamma heavy chain disease, which comes from a cut off gamma chain that lacks light-chain binding sites
LPL treatment update:
CXCR4 mutations also of interest and only seen infrequently in some other B-cell lymphomas, not generally used for diagnostic purposes (best with targeted NGS or sanger bc >40 variants)
- present in up to 40% of cases
- in contrast to MYD88, subclonal
- mutually exclusive with 6q deletions
- assoc c less adenopathy, nonsense mutations assoc c inc BM dz, IgM levels and/or sympromatic hyperviscocity)
MYD88 mutation only: favor ibrutinib monotherapy
• MYD88 and CXCR4 mutation: combination of anti-CD20 antibody with alkylating agent or BTK inhibitor
• Absence of MYD88 and CXCR4 mutation: have a worse px, and a lower response to ibrutinib, MYD88 cases may be a different type of disease altogether, alkylating agent and anti-CD20 or proteosome inhibitors
• New developments; anti-CXCR4 monoclonal antibody, small molecular inhibitors of CXCR4,
Treon has suggested looking for mutation in the TIR region of MYD88 in casese that otherwise appear to be unmutated bc they also respond to ibrutinib

Lymphoplasmacytic lymphoma
2017 Waldenstroms definition need LPL in BM and IgM monoclonal gammopathy of any concentration


LPL Dutcher body

Many diseases have MYD88 mutation - not specific

lgM Monoclonal gammopathy of undetermined significance
(IgM MGUS)
• In 2017 WHO, own distinct entity
- Must distinguish IgM MGUS that may be MYD88 mutated from non-IgM MGUS which does not have MYD88 mutations
• Recognized to be more akin to LPL than to myeloma (unlike IgG and IgA MGUS, which is more similar to plasma cell neoplasms)
• Much less prevalent than IgG and IgA MGUS
_____________________________________
Diagnostic criteria
• Serum IgM monoclonal protein <30 g/L
• Bone marrow lymphoplasmacytic infiltrate <10%
• No evidence of CRAB, constitutional symptoms, hyperviscosity, etc. that could be attributed to lymphoma (lymphadenopathy [LAD])
_____________________________________
Micro: plasma cells and plasmacytoid lymphocytes <10% of the marrow cellularity
- mildly increased plasma cells, usually well-differentiated, clonality can be difficult to detect by IHC (flow cytometry may be helpful in this situation)
- May be impossible to distinguish bwt reactive plasmacuytosis and MGUS
- may appear completely normal!
- requires correlation with clinical data
_____________________________________
IHC: clonal B-cells seen in 75% of IgM MGUS by flow, with a phenotype similar to LPL, plasma cells CD56-
_____________________________________
Genes: Approximately 50% of cases harbor MYD88 L265P mutation; 20% have CXCR4 mutations (also in 30% of LPL, but not in IgG/A MGUS)
_____________________________________
Px: risk of progression is 1.5% per year, goes to LPL/WM, amyloidosis, or other lymphoma
- compare to Non-Igm MGUS, which has1% progression per year to MM, plasmacytoma, amyloidosis
- light chain MGUS has 0.3%/year progession to MM/amyloidsis
_____________________________________
Key tips
• Clonal B-cell component and/or clonal plasma cell component may be difficult to detect
– Low numbers to being with
– Within a polyclonal background
• Assure adequate sensitivity of your MYD88 assay
Evaluation of M protein in BM

MGUS risk of progression [14]

IgG4 related disease
• Lymphoplasmacytic infiltrate and fibrosis
• IgG4/IgG ratio >0.4
• >100 polyclonal plasma cells/hpf
• Eosinophils
• Follicular hyperplasia
• PB eosinophilia, hypergammaglobulinemia, ↑il-2 receptor, hypocomplementemia
• Serum IgG4 levels elevated, but normal in 30%
• Autoimmune pancreatitis, LN and orbital disease
• Other diseases such as IBD and Castleman's disease can have inc IgG4+ plasma cells, but are NOT IgG4 related disease
Tx: steroids
Heavy Chain Diseases (HCD)
Group of diseases where only heavy (H) chain of immunoglobulin produced; includes: IgA (alpha HCD), IgG (gamma HCD), IgM (mu HCD)
- heavy chains incomplete and of various sizes, thus won't be able to fully assemble
-- b/c chains are variably-sized, may throw off electrophoresis results (need to run immunoelectrophoresis or do immunofixation)
MC is alpha HCD - variant of extranodal marginal zone lymphoma of MALT;
occurs in young adults (10-30 yo) from Mediterranean and affects the GI tract, known as immunoproliferative small intestine disease (IPSID) thus causing sx of diarrhea, fever, abd pain, hypoCa+ and wasting
- may result from chronic infx c C. Jejuni
Genes: deletions in IGHA gene present that cause defective heavy chain protein that cannot bind light chain to make a complete immunoglobulin
- does not have t(11;18)
Px: may completely remit c abx; most progress to DLBCL; some anthracycline-chemo tx may help
Mu heavy chain disease
Production of defective my heavy chain lacking a variable region
- routine protein electrophoresis is usually normal (need to do immunoelectrophoresis); 1/2 have Bence-Jones proteins (kappa chains) in urine
Resembles CLL (clinically and on histo); super-rare dz
- can distinguish from CLL bc usually has HSmegaly and no LN involvement
BM has infiltrate of plasma cells c vacuoles mixed c small round lymphs; LN usually not involved
- affected B-cells have monoclonal cytoplasmic mu HC, but otherwise have normal IHC profile ([-]CD5/10)
Ig genes clonally rearranged and have high levels of somatic hypermutation
- gamma heavy chain deletions cause production of a defective heavy chain protein that cannot bind light chain to complete the Ig molecule
-- large amts of DNA insert into the CH1 region and there's VH gene deletions
BM and other organs not usually involved, though can be in lungs
Will see hypogammaglobulinemia on SPE b/c HC assembly defective
Look for infiltration of ab-producing cells in the lamina propria of GI; may progress to DLBCL
Usually indolent
Gamma HCD
Llooks like lymphoplasmacytic lymphoma, is called Franklin H chain disease
Rare dz c protein produced by lymphs, plasmacytoid lymphs and plasma cells which has no light chain (thus an incomplete free-floating molecule)
- can involve any lymphoid tissue; may see these ab-producing cells in PB
May present c a variety of autoimmune/rheumatologic dzs (Sjogrens, RA, MG, cutaneous SLE)
- no lytic lesions or amyloid deposition, but BM involed in ~50%
LN may resemble HL or AITL, c mix of ab-producing cells and histios, eos, and immunoblasts
- affected cells have IgG without light chains inside them, and otherwise have normal IHC markers
Variable px, from rapidly progressive to indolent (survive 1-8 years)
- low-grade lymphoplasmacytic infiltrates in LN respond to non-anthracycline chemo; may respond to rituximab
Plasma cell neoplasms
Immunoglobulin tests
Protein Electrophoresis
- good screening method
Immunofixation (monoclonal protein analysis)
- good to document the type of light chains present
Quantitative immunoglobulins
- measures the amount of each type of protein
Serum free light chain ratio
- sensitive method that measures amount of proteins not bound to heavy chains
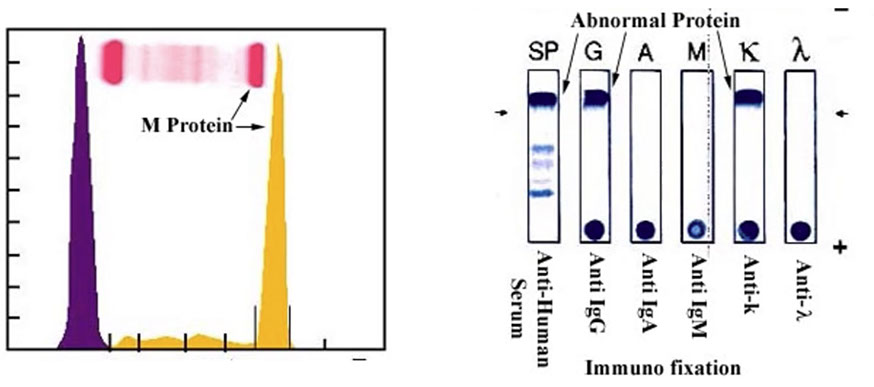
Protein electrophoresis - Normal [14]. Low level monoclonal proteins can be missed with SPEP/UPEP alone; some abnormal Ig can migrate in beta region; other tests are more sensitive (eg free light chains, IFIX)

Abnormal Protein Electrophoresis [14]. On the left, see normal albumin and a and b peaks, then abnormal M protein spike. See on the right that there are IgG heavy chains and kappa light chains
Non-lgM monoclonal gammopathy of undetermined
significance (Non-IgM MGUS)
Dx criteria:
- serum IgG/IgA protein <3q/dl
- clonal plasma cells <10%
- no CRAB or amyloidosis
_____________________________________
- MGUS is the presence of monoclonal protein without evidence of lymphoma, myeloma, or other related disorder
- "Benign" gammopathy
- usually an incidental findings
- 1% of the population >50 yo, 3% >70 yo
Px: 25% of patients will go on to develop overt myeloma, lymphoma, etc
- avg time to progression = 10 years
- remainder of pts remain symptomatic
- pts require lifelong follow-up
MGUS risk of progression [14]
Evaluation of M protein in BM
Light Chain Monoclonal Gammopathy of Undetermined Significance (MGUS)
Dx Criteria:
- No heavy chain on immunofixartion
- No CRAB or amyloidosis
- clonal bone marrow plasma cells <10%
- urine monoclonal protein <500 mg/24hr
__________________________________________


Plasma cell myeloma
aka Multiple Myeloma
~1% of malignant cancers; ~15% of heme cancers; ~20% deaths from heme malignancies
- MC in men (~1.5:1) and blacks (2:1)
-- not seen in children (90% after 50 yo; avg age 70 yo); ~4x greater risk c + FamHx
May be caused by chronic infx/dz/toxin exposure
- BM typically involved
Monoclonal (M)-protein in serum or urine
- usually both heavy and light chain (85%)
- usually IgG or IgA, rarely IgM, IgD or IgE
- 15% light chain only (Bence Jones protein)
- rarely nonsecretory
_____________________________________
Dx criteria
Biopsy proven plasmacytoma and/or >10% clonal plasma cells in bone marrow AND at least one of:
- Symptomatic plasma cell myeloma:
- hyperCalcemia, Renal problems, Anemia, Bone lesions (CRAB)
- Myeloma defining events: >60% plasma cells, FLC ratio >100, two or more lytic lesions >5mm on MRI
*** SLIM CRAB*** Sixty (%pcs) LI-ght chain ratio >100, MRI >1 lytic lesion ***
_____________________________________
Micro: increased plasma cells
- plasma cells may be morphologically unremarkable ("Marshalko type")
- may be multinucleated (binucleate plasma cells can be seen in reactive conditions, although the more of these you see the more you think that it is neoplastic)
- may have visible nucleoli
- variant morphologies exist (lymphoplasmacytic, cleaved, anaplastic, blastic, etc.)
_____________________________________Flow cytometry: usually underestimates plasma cell numbers
- CD45 dim, CD138 positive gating
- CD19 negative
- CD56+, normal plasma cells are CD56-
- may have myeloid antigens (CD13, CD33, CD117, etc.)
- sIg neg, but monotypic cIg light chain
_____________________________________
IHC: CD138, kappa and lambda may be useful to document clonality
- numbers often greater than seen in H&E alone
- clonal populations may be missed if small
- normal plasma cells: Kappa > Lambda (2-3:1)
_____________________________________
Cytogenetics:
by flow cytometric ploidy analysis and FISH, cytogenetic abnormalities nearly universal in myeloma
- classical cytogenetics - only 30% abnormal karyotypes
-- probably bc low proliferative rate of plasma cells in culture
- cryptic abnormalities
-- FISH is the gold standard
50-60% of cases have a translocation involving IGH (14q32)
-- several partner genes described
- usually non-hyperdiploid
40-50% of cases lack IGH translocations
-- usually hyperdiploid
Dysregulate Cyclin D1, D2, or D3
***anything 14 and above is poor!!!***
t(11;14)(q13;q32)
- seen by classical cytogenetics, if abnormal metaphases present
- many cases detected only by FISH or molecular techniques
- neutral or slightly better prognosis
- assoc c lymphoplasmacytic morphology and CD20 expression
- Cyclin D1+ by IHC
t(4;14)(p16.3;q32)
cryptic translocation
- requires molecular techniques
- adverse prognosis
- assoc in some cases with "plasmablastic"morphology
- assoc c FGFR3+ by IHC
t(14;16)(q32;q23)
May be cryptic by metaphase cytogenetics
- requires FISH or other molecular technique
- assoc c poor px
Other cytogenetic Abnormalities in Plasma cell myeloma [14]
- Gain of 1q (adverse px)
- monosomy 13 or del(13q) (adverse px if seen on karyotype)
- 17p abnormalities (adverse px; p53+ by IHC)
- many others
_____________________________________
"High risk" myeloma
- 1q21 gain, 17p13 loss, t(4;14), t(14;16)
"Standard risk myeloma"
- all others including t(11;14) and hyperdiploidy
_____________________________________
Tx:
Minimal Residual Myeloma
- allele specific Oligonucleotide-qPCR
- NGS
- High-sensitivity flow cytometry
_____________________________________
Px: poor
- median survival apprximately 6 years
- other new agents may provide additional benefit
Russell bodies

Morphologically unremarkable (Marshalko) plasma cells
Plasma cell w prominent pale nucleolus should make you very suspicious [14]

myeloma trnslocations. t(11;14) is the same one seen in mantle cell
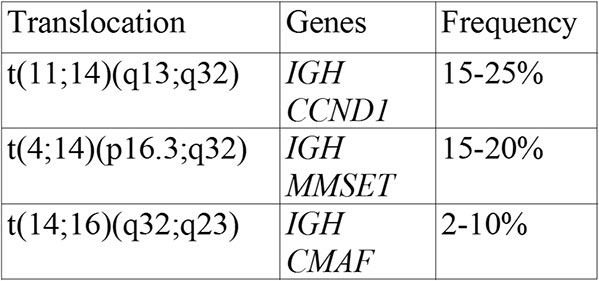
Example of myeloma with t(11;14) sometimes called the small cell variant of plasma cell myeloma, may be easily confused with a lymphoid process

Myeloma with t(4;14) that looks blastic


IgM Myeloma
Rare; less than 1% of cases of myeloma
• Plasma cell proliferation is usual; a subset of cases may show a striking lymphoplasmacytic proliferation as well
• Co-expression of CD20 and/or CD19 on the plasma cells in some cases (complicates the distinction from LPL)
• Cyclin D1 positive (40-50%), MYD88 mutation negative.
IgM Myeloma


Plasma cell myeloma variants
Smoldering (asymptomatic) plasma cell myeloma
Have dx criteria for MM (+M-protein), but no CRAB
- is similar to MGUS, but is more likely to progress to full MM (10% risk per annum in 1st 5 yrs, 3% per annum yr 6-10, 1% per annum thereafter; also, 8% of MM are initially asymptomatic)
_____________________________________
Dx Criteria
- IgG or IgA protein >3 g/dl in serum AND/OR urine >500mg/24hrs AND/OR 10-60% plasma cells (if>60% plasma cells is considered a "myeloma-defining event" and is not "smoldering")
- No CRAB, "Myeloma-defining event" (MDE), or amyloidosis
- risk of myeloma or amyloidosis: 10% per year for first 5 years
_____________________________________
70% of pts c MM have lytic lesions
97% have serum/urine M-protein (50% IgG, 20% IgA,
20% LC); 3% non-secretory
- in kidney, tubular injury causes hypercalcemia +/- hyperuricemia; AL amyloidosis assoc c delta-light chains (LCs); LC deposition dz MCC by kappa-LCs
- can also have myeloma cast nephropathy
Paraproteins MC isotype (Heavy Chain [HC]) is IgG (>1/2), IgA (1/5), none-LC only (1/5), IgD (1/50), biclonal (1/50), or IgE (1/50)
- the MC idiotype (LC) is kappa
- non-secretory myeloma in 1/20
- Russell bodies caused by accumulation of Ig's in endoplasmic reticulum in plasma cells
Non-secretory myeloma
Pts have sx of CRAB, but to a lesser degree
- will see inc plasma cells in BM, which have cytoplasmic M-protein
- includes Durie-Salmon dz, stage 1
Plasma Cell Leukemia (PCL)
>20% of WBCs in PB are monoclonal plasma cells, and can be seen in other organs; 1/2 arise de novo, presenting abruptly c aggressive course
- seen in 2-5% of cases of MM as a late event in the disease progression; plasma cells in PB may resemble plasmacytoid lymphs, c little cytoplasm and small size
Do not have lytic lesions / bone pain as much as MM, and lymphadenopathy / organomegaly and renal failure are more common
Clonal plasma cells constitute >20% of total leukocytes in blood or absolute count is >2
_____________________________________
Micro:
In PB, may be difficult to appreciate plasma cells, often need flow cytometry to distinguish from lymphoid leukemia (CD138+, monotypic cIg, CD19-, CD56 may be + or -)
On aspirate, plasma cells can vary from few to up to 90% of cells, and can vary in morphology and degree of differentiation
- plasma cell % may be low due to poor aspirate
Mature plasma cells have round, eccentric nucleus c "clock face" or "spoke wheel" chromatin and perinuclear hof (clearing)
Immature plasma cells have higher N:C, prominent nucleoli, and more dispersed chromatin (more likely to be malignant c immature plasma cell proliferations)
Interesting cell types which can be seen in reactive or malignant conditions:
- Mott/Morula cells have blue-white grape-like cytoplasmic bodies
- Flame cells: reddish cells c glycogen-rich IgA
- Gaucher-like cells have overstuffed fibrils
Renal tubules may show accumulation of reddish material, made of Bence Jones protein (causes renal damage)
_____________________________________
IHC: (+) CD38/79a/138 / 117 (in 1/3) / 20 / 52 / 10, cytoplasmic Ig (kappa or lambda), PCA1, myeloid markers in up to 1/3 (CD117/13/33/11b/15), up to 1/2 are CD10+; sometimes CD30+ or EMA+
- neg: CD19 / 20 / 45 / 43 (var) / 56, PAX5, surface Ig
- CyclinD1 positivity assoc c t(11;14)(q13;q32) c CCND1 gene and presence of lymphoplasmacytic cells
_____________________________________
Genes:
Ig HC and LC genes clonally rearranged, and lots of IGHV gene somatic hypermutation which comes from post-germinal center B-cells
1/3 of pts have cytogenetic abnormalities; 90% have FISH abnormalities
- up to 70% have IGH@ translocation on chr 14q32, the major 2 involving cyclinD1 (11q13; in up to 18%), and FGFR3/MMSET (4p16.3, in up to 15%)
-- a wide assortment of abnormalities can otherwise be seen
Classified into 8 groups based on translocations (T) and cyclin (C) D expression (TC groups)
- each group can represent a different entity c different tx's
-- MC groups are the D1 group (34%, no gene assoc, good px); D2 (17%, no gene assoc, unknown px), 11q13 group (16%, CCND1 gene, good px) and 4p16 group (15%, FGFR3/MMSET gene, poor px)
- other grouping systems using molecular genetics is similar and breaks up MM into 7 groups
Monosomy / del chr 13 (13q14) is an early event in pathogenesis, poorly understood
- found in 50% of cases by FISH
K-/NRAS activating mutations in up to 40% and may cause progression of MGUS
_____________________________________
Staging (Durie and Salmon system)
I: Low M-protein, absent/solitary lytic bone lesions, no CRAB, normal Ig levels
II: bwt I and III
III: High M-protein, multiple lytic lesions, severe CRAB
_____________________________________
Tx:
_____________________________________
Px: most aggressive plasma cell neoplasm
- worst have abnormalities by conventional cytogenetics, also if high B2 microglobulin levels, high plasma cell labelling index, high stage
- most imp px indicators: t(4:14), MAF translocations [t(14;16) and t(14/20)], del 17p/TP53, and inc B2 macroglobulin in serum
Plasma cell leukemia



Plasma cell leukemia, the plasma cells may be difficult to recognize as plasma cells in the PB [14]

Plasmacytomas
solitary plasmacytoma of bone; Extraosseous [extramedullary] plasmacytoma
- <5% of all plasma cell dyscrasias
Solitary Plasmacytoma of Bone
Localized bone tumor of monoclonal plasma cells, without any other radiographically evident lesions, usually causing bone pain
- 3-5% of plasma cell neoplasms; slightly more prevalent in men
-- spine > ribs > skull > pelvis > femur (sites c active hematopoiesis)
~75% have inc serum M-protein; no CRAB
_____________________________________
Diagnostic criteria:
- Biopsy proven solitary lesion of clonal plasma cells
- Normal imaging (skeletal survey, MRI, or CT) except for the lesion in question
- No CRAB or amyloidosis
- Normal bone marrow (no clonal plasma cells)
_____________________________________
Tx
- 90% can be controlled with local radiotherapy
- M protein can be followed to track therapeutic response/ recurrence
_____________________________________
Px
Mean survival: 10 years
- 1/3 of pts disease free for 10 yrs, but usually progresses to MM in 2-3 years
-Adverse px factors are osteopenia and low levels of other Igs, which suggest occult myeloma
plasmacytoma of rib [14]
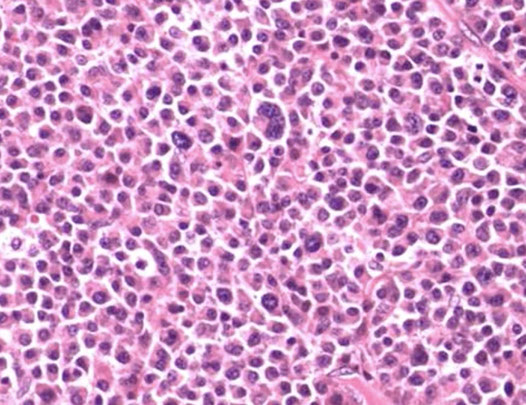
Solitary Plasmacytoma with Minimal BM Involvement
Diagnostic criteria:
- Biopsy proven solitary lesion of clonal plasma cells
- normal imaging (skeletal survey, MRI or CT) except for the lesion in question
- No CRAB or amyloidosis
- <10% clonal plasma cells in BM
__________________________________________
Extraosseous (extramedullary) plasmacytoma
>5% plasma cell tumors.
May have common origin w/ extramedullary plasmacytoma and marginal zone lymphoma
Usually (>75%) occur in upper respiratory tract, and met to cervical lymph nodes in 15%
- 20% have M protein (MC IgA)
DDx:
Reactive lesions or lymphomas w/ lots of plasma cells - but these have POLYclonal kappa/lambda light chain staining
- difficult to distinguish from lymphoma w/ advanced differentiation of plasma cells
-- immune-/plasmablastic cell lymphomas, esp. marginal zone lymphoma of MALT
Tx
Radiotherapy
- 1/4 pts get local recurrence/mets; 15% turn into MM
- 70% 10-yr disease free survival
-- indolent cases suggest closer relation to MALT lymphoma than MM
Px: most do not have detectable M protein, and don't get MM
Monoclonal Immunoglobulin Deposition Diseases
Monoclonal Immunoglobulin Deposition Diseases (MIDD) have visceral/soft tissue deposition of Igs, resulting in organ dysfunction
Usually have underlying plasma cell / (or rarely) lymphoplasmacytic neoplasm
- Ig deposition occurs before underlying tumors are significantly large
2 main categories are: 1) Primary Amyloidosis; and 2) LCDD
Primary Amyloidosis
Caused by underlying neoplasm that secretes abnormal Ig LCs which deposit in organs, causing dysfunction, and form Beta-pleated sheets, linear non-branching 7.5-10 nm fibrils
AL is the most common form of amyloid (up to 2/3)
- deposition of Ig light chains
~20% of AL patients have myeloma
- has characteristic birefringence with Congo red
Avg age at dx:64; MC in males
- early signs of dz are purpura, neuropathy, bleeding and carpal tunnel syndrome
-- commonly see congestive heart failure (CHF), cardiomyomapthy, nephrotic syndrome, chronic renal failure (CRF), malabsorption, hepatomegaly, pulmonary involvement, soft tissue and joint involvement, Factor X deficiency in coagulation tests
Most pts have criteria for MGUS, but 20% have underlying myeloma
- will find abnormal proteins in 99% using immunofixation and free light chain ratio
_____________________________________
Micro:
Waxy, amorphous, homogenous material
- may be seen in vessels, other extracellular deposits in BM or extramedullary tissues
- plasma cells may be present, but BM should contain <10% plasma cells
Macrophage eat up Ig light chains and spit them out into ECM
Grossly will appear waxy or like porcelain
May be diagnosed with fat pad aspirate, rectal biopsy, heart biopsy, BM bx, other organ involvement (shouidl bioppsy the site the clinician thinks is involved
__________________________________________
Rare variants [14]
- Heavy chain amyloid (rare, may be a, g, or u)
- non-amyloid heavy and light chain deposition (rare, deposition of heavy, light, or both chains into non-amyloid structure, Congo red negative)
__________________________________________
Stains:
Congo red
Thioflavin S (fluorescent scope needed)
IHC for Ig light chains, P component (found in most forms of amyloid), Amyloid A
Has become more and more routine to analyze amyloid with mass spec
- becoming the gold standard for amyloid typing
- IHC classification has well-known errors
- Serious consequences!!!
__________________________________________
Tx: bone marrow transplant
__________________________________________
Px: poor, 1-3 years
- px even worse in pts with MM
Many types of proteins can cause amyloid deposits [14]

Pulmonary amyloidoma [14]
Left - congo red, showing orangish color, middle is apple-green birefringence, right is the P component



Light Chain and Heavy Chain Deposition Diseases
LCDD and HCDD
Secretion of LC or HC that deposit in tissue (similar to primary amyloidosis) but do not for amyloid-beta-sheets, bind Congo red, or have amyloid P-component
- assoc c MGUS or (more often) MM
- commonly involves kidneys, but also heart, liver, nerves, vessels and joints causing dysfunction
-- see amorphous, refractile red stuff in kidney bx
M-protein production driven by genetic alterations (mutations, deletions)
- in LCDD VkIV LC is overproduced; in HCDD the CH1 constant domain altered causing a product c aa's to be secreted that have tendency to deposit in tissues
In LCDD, see granular, linear kappa chains deposited along BM of tubules
- prevalent kappa light chains (vs lambda light chains in primary amyloidosis)
Px: 4 yr avg survival; depending on age, underlying neoplasm
Plasma cell neoplasms with associated paraneoplastic syndrome
- POEMS syndome and TEMPI syndrome
Osteosclerotic myeloma (POEMS syndrome)
POEMS = Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal gammopathy and Skin changes
Plasma cell neoplasm c fibrosis and osteosclerotic changes in bony trabeculae
- usually c lymph nodes affected
- may be considered a plasma cell variant of Castleman's disease
CRITERIA
Mandatory criteria
- Polyneuropathy (typically demyelinating)
- Monoclonal plasma cell proliferative disorder
Major criteria (>1 needed)
- Castleman disease
- Osteosclerotic bone lesions
- VEGF elevation
Minor criteria (>1 needed)
- Organomegaly
- Endocrinopathy
- Skin changes
- Papilledema
- Thrombocytosis
- Extravascular volume overload
- most pts don't have all the sx, but instead just a progressive neuropathy
-- skin changes in 2/3; organomegaly in 50%
- ~80% have inc serum M-protein (IgG/A lambda); 50% c urine M-protein
-- CRAB is rare
Rare; 1-2% of plasma cell disorders; MC in Japan (?)
- pts commonly have imbalance of inflammatory mediators; may be due to production of VEGF by tumor cells
- when assoc c Castleman dz, think HHV-8
Characteristic lesion is osteosclerotic plasmacytoma (single or multiple)
- thick trabecular bone c paratrabecular fibrosis that entraps and distorts plasma cells
- will not see inc # of plasma cells
- if lymphadenopathy present, assoc c plasma cell variant of Castleman dz
Mean survival is over 14 yrs
- death by cardiorespiratory failure and infx
Skin lesions typical in POEMS
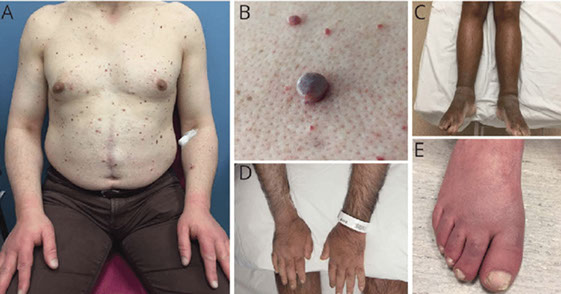
POEMS criteria
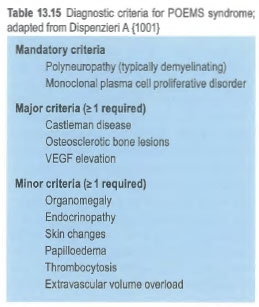
TEMPI Syndrome
- Telangiectasias, elevated Erythropoietin and erythrocytosis, Monoclonal gammopathy, Perinephric fluid collection, and Intrapulmonary shunting.
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma)
B-cell cancer defined by their recapitulation of Peyer's patch lymphoid tissue and with the major neoplastic compoenent originating from the marginal zone compartment (the degree of Peyer's patch varies)
- occur at mucosal and at certain non-mucosal sites
- initial bx may come from a LN bx
- may arise at sites without normal organized lymphoid tissue and are associated with antigenic stimulation and or autoimmune disorders -- arise at sites of "acquired" MALT (maybe infection, self antigens???)
assoc c H. pylori (although the % is reportedly decreasing, 23-43% of cases)
- benign acquired MALT (gastritis) --> lymphoma
- the T-cells are strain-specific but the neoplastic B-cells are autoreactive
- more recent suggestion of direct effect of H pylori on B-cells including oncogenic effect of CagA
- autoimmune disorders are also important
- if occurring in extranodal tissue that has cells which look like marginal zone (centrocyte-like) cells, monocytoid cells, small lymphs, scattered immunoblasts and centroblasts
- up to 8% of all B-cell lymphomas; 1/2 primary gastric lymphomas
- 7th decade, F>M; MC in GI tract (stomach 38%, where33%involve the stomach only, then salivary gland, skin, conjunctiva)
Site-related differences are important [9]
- Histopathologic differences - frequency of: plasmacytic differentiation (more common in primary cutaneous and thyroid MALT lymphomas), monocytoid-appearing cells (very common is salivary, but not common in skin MALT lymphomas), lymphoepithelial lesions (not common in the breast and skin)
- Biology: Different frequency of varied cytogenetic abnormalities, different frequency of varied mutations (for example, orbital/ocular adnexal MALT lmyphomas reported to have slightly more frequent MYD88 L265P mutations, where it also has been reported to be an adverse progrnostic indicator), and different IGVH gene usage
- Have different clinical associations and outcomes based on the site
ImmunoProliferative Small Interstinal Disease (IPSID) is an alpha heavy chain disease seen in Italy, South Adrica, and Middle east and is assoc c Campylobacter jejuni
- Chlamydia psittaci, C. pneumoniae, and C. trachomatis assoc c adnexal (lacrimal gland /orbital) MALT lymphoma
- B burgdorferi assoc c cutaneous MALT lymphoma
- Sjogrens (SS) with Hepatitis C virus= salivary MALToma
-- SS and lymphoepithelial sialadenitis (LESA) = overt lymphoma
- Hashimotos = thyroid MALToma
- Achromobacter xylosoxidans in pulmonary MALT lymphoma
Micro: lymphoma cells go circumferential to reactive B-cell follicles outside of mantle zone and in marginal zone
Lymphoepithelial lesions: infiltration of neoplastic cells (>3) into recogniozable epithelial structures often with resulting epithelial alterations (oxyphilic change as in Hashimotos)
- lymphoepithelial lesions = aggregates of 3+ marginal zone cells c gland epithelial destruction
- may impart a starry sky pattern around the follicles
- can have Dutcher bodies
- are NOT specific: seen in florid gastritis, and other B- and T-cell lymphomas
- not a required finding for dx, MALT lymphomas can arise at sites without epithelial structures, and are very infrequent in some MALT lymphomas
Important!!! MALT lymphoms are NOT diagnosed when there are numerous large and transformed cells!!!
IHC: (+) IgM>IgA/IgG, CD20,79a,43+/-,11c+/- (weak), MNDA, IRTA
- negative: CD5 (+ in <1%), 10, 23,
- plasma cell differentiation in 1/3 of MALT lymphomas
CD43 (T-cells, myeloid) -- CD43+ B-cell proliferation supports diagnosis of B-cell lymphoma but many MALT lymphomas are negative
- CD43 benign B-cells can be small normal population including tonsilar intraepithelial B-cells, some tonsilar/appendiceal marginal zone hyperplasias, at least in children (can also be lambda light chain class restricted!), and salivary gland (such as lymphoepithelial sialadenitis)
- should NOT diagnose something as lymphoma, especially at a mucosal site, just because it is CD43+
Epstein-Barr virus (EBV)+ marginal zone lympoma
- Described in MALT lymphomas in the post-transplant setting and in other settings with immune suppression including just old age
- most often extranodal (cutaneous) with prominent plasmacytoid differentiation
- different than with other inciting agents as EBV is actually in the neoplastic cells
- clinically indolent with good response to reduction of immunosuppression, localized therapy, or immunochemotherapy [23]
Genes: many MALT lymhphomas have cytogenetic abnormalities that can be documented using FISH, although most are diagnosed on the basis of histopathology and phenotype, sometimes molecular clonality studies can be helpful though need to be careful because monoclonality can be seen in some cases of gastritis, or following disappearance of gastric MALT lymphomas with H pylori eradication
- trisomies -- 3, 18, 12 (not specific)
Translocations involving MALT1 gene:
- t(11;18)(q21;q21), with BIRC3 (aka API2)
- t(14;18)(q32;q21), with IGH and MALT1 gene
t(1;14)(p22;q32), involves IGH and BCL10 gene
t(3;14)(p14.1;q32) with FOXP1 and IGH
-- t(11;14) results in a fusion gene, where BIRC3 (aka API2) fused to C-terminal sequences of MALT1, promoting cell survival and proliferation via activation of NF-kappaB
-- t(1;14)(p22;q32) and t(14;18)(q32;q21) similarly deregulate BCL10 and MALT1, respectively, by juxtaposition next to IGH enhancer
- t(11:18) tumors are usually resistant to H pylori tx
- t(14;18) IgH-MALT1 seen in MALT lymphoma in ocular adnexa / orbital and salivary gland lesions
- t(3;14) IgH FOXP1 in thyroid, ocular and skin MALT
MALT lymphoma does NOT have CCND1 translocations, rare cases with BCL2 (HCV+) or BCL6 translocation
DDx: Most common problem is an atypical reactive lesion wotj or without follicular colonization (neoplastic infiltration of follicular centers) (chronic gastritis, LIP of lung, cutaneous lymphoid hyperplasia), and the most helpful finding is the demonstration of monotypic light chain expression (be careful bc subepithelial plasma cells can be monotypic) - CD43 expression favors lymphoma, but it is not specific, and plasma cells can also be CD43+
- some cases have sheets of plasma cells (ddx plasmacytoma)
- intrafollicular transformation does NOT equal DLBCL
- may have intrafollicular plasma cell differentiation by MALT lymphoma
MALT vs DLBCL
- today diagnosis of MALT means predominantly different types of small non-transformed lymphocytes +/- plasma cells
- precise criteria for how many large transformed cells are not well defined
- no established criteria for grading MALT lymphomas, "increased" transformed cells found by some, but not by all, to be adverse prognostic indicator in gastric MALT
MALT lymphomas can be difficult to diagnose morphologically because of the non-specificity of many of the morphologic features, the absence of one of more classic features and the nature of the small biopsies in many cases
-- Major ddx: Lymphoid hyperplasia (gstritis), other small B-cell lymphoid neoplasms, DLBCL, and T-cell lymphoma
Flow cytometry: helpful if CD5- CD10- monotypic B-cells but false negative results are not unusual bc of admixed reactive B-cells
- a signficant minority have monotypic plasma cells so kappa and lambda stains are worth a try , but many MALT lymphomas have polytypic plasma cells and some T-cell lymphomas (TFH cell lymphomas) have monotypic plasma cells
- presence of a very B-cell rich proliferation at an extranodal site will favor the diagnosis of lymphoma and if the issue is the type of lymphoma will support the diagnosis of a B-cell lymphoma, but.... some MALT lymphomas have many admixed T-cells, and cases repord with florid T-follicular helper cell hyperplasia in a soft tissue MALT lymhpoma
- CD20 may underestimate the number of B-cells in lymphomas with plasmacytic differentiation so CD79a stain may be useful
Most cases are distinguishable from CLL/SLL, FL and MCL based on paraffin section stains showing absence of CD5, CD10, BCL6 and cyclin D1 expression but...
- the phenotype of a MALT/MZL might suggest a different type of ML (and vice versa)
-- CD5+ cases (more disseminated/ aggressive/non-gastric)
-- CD10+ cases (very rare, BCL6-)
- should NOT be cyclin D1+
Tx: possible remission c H pylori tx, many respond to antibiotic induced eradication of H pylori (median to remission is 4-5 months, some consider failure at 12 months)
- CR in 75% of cases,
- 5-22% relapse often related to H pylori reinfection -- need long f/u
- 15-33% response in H pylori negative cases
- sensitive to rads
Px: indolent and slow to disseminate
- overall 5-year survival 80-95% for gastric MALT lymphoma with very few patients dying of lymphoma and overall 72% 10 year survival
- variable successes with some other MALT lymphomas
MALT lymphomas usually localized; other small B-cell lymphomas usually disseminated
- about 1/3 of MALT lymphomas are disseminate with BM involved in about 10-20%
- gastric MALT: 88% stage1, 7% BM+
Therapeutic appreoach is often very different
- salivary gland MALT lymphomas often have been treated with surgery alone (becaues dx as benign in the past)
- cutaneous MALT lymphomas often treated with localized therapy (surgery and/or radiation
Components of a MALT lyphoma
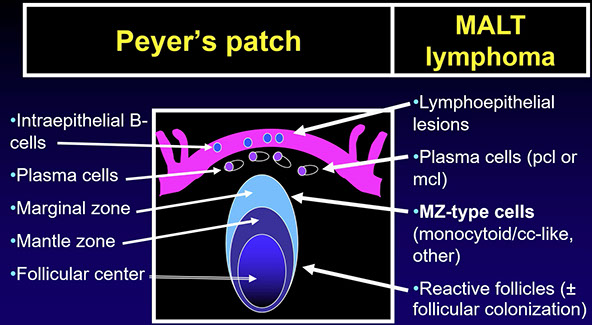
MALT lymphoma components, low power

MALT lymphoma with predominantly small lymphocytes around outside invading into what looks like a residual GC in the middle with larger
- has numerous small lymphocytes including many of apparent marginal zone type (cells with more aundant / monocytoid cytoplasm and often somewhat irregular "centrocyte-like" nuclei

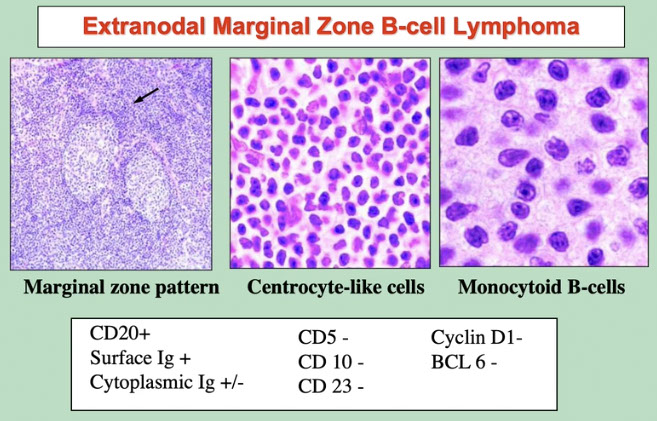
Surrounds residual reactive follicles
Has immunophenotype of exclusion

Characteristic lymphoepithelial lesions in gastric MALT lymphoma where neoplstic cells invade glandular epithelium [4]
MALT1 gene

How to get to the diagnosis of a MALT lymphoma

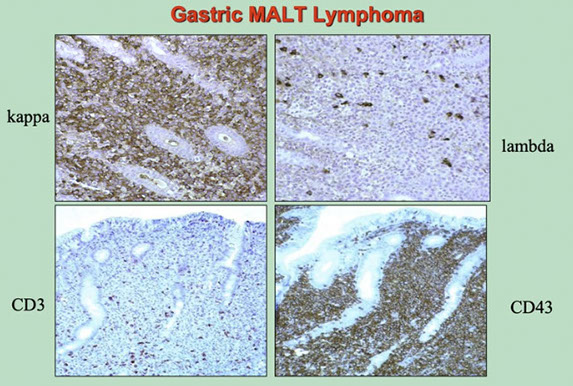
Typical case of gastric MALT lymphoma showing kappa-restriction, and CD43+ versus a small number of CD3+ T-cells. CD43+ favors lymphoma over reactive, as does the monoclonal Ig
High-power view of the plasmacytoid cells (left) which are on the top of the epithelium on the picture to the left, and the monocytoid cells (right image) seen under the plasmacytoid cells in the image to the left

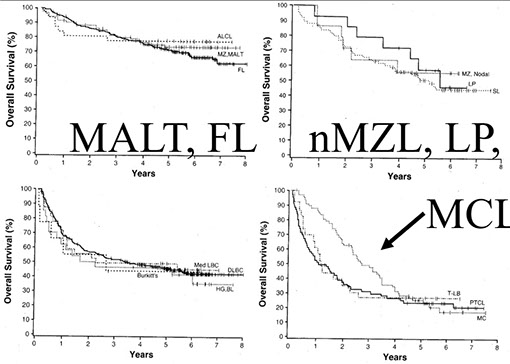
Survival curves of MALT, FL --- nMZL, LP, --- Burkitts, DLBCL, ---- and MCL
Survival outcomes in pts with MALT lymphoma based on site
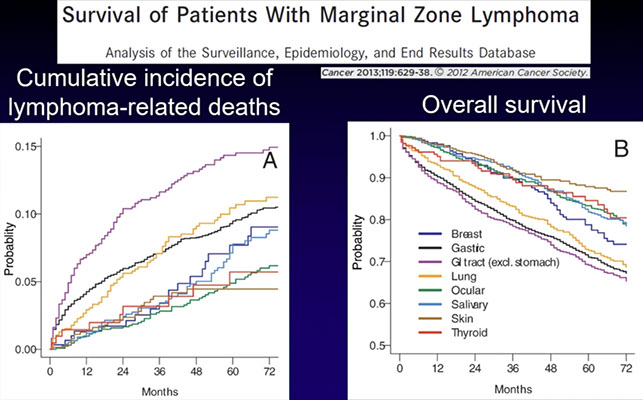
MALT lymphoma lymphoepithelial lesions: infiltration of neoplastic cells (>3) into recogniozable epithelial structures often with resulting epithelial alterations (oxyphilic change as in Hashimotos); cytokeratins are generally not necessary for diagnosis
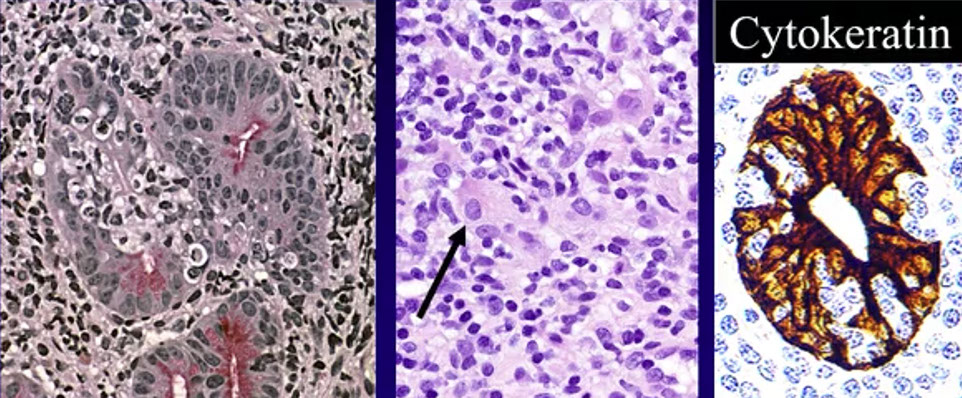

MALT lymphoma with superficial portion with plasmacytoid differentiation strongly expressing Lambda, which is distinct from the deep part that is more monocytoid

CD21 stain for FDC highlights extensive follicular structures colonized by the lymphoma

MALT lymphoma see the BCL6+, BCL2- reactive GC remnants, associated with follcular colonization in marginal zone lymphomas (believed to represent neoplastic cells with upregulation of BCL6)
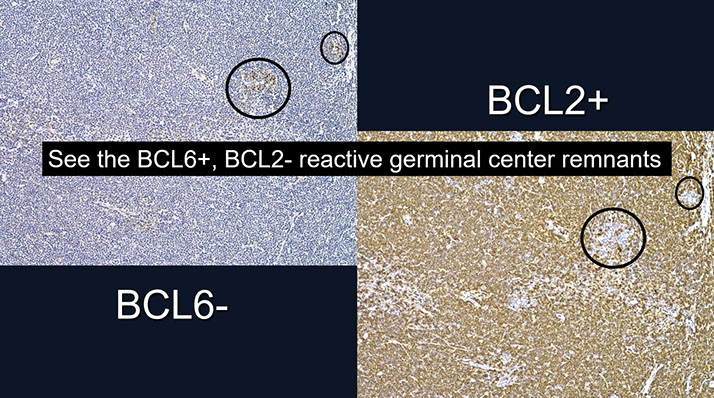

Same case as surrounding iamges with plasmacytic differentiation as the image to the left and above with plasma cell differentiation in the top layer negative for CD20 and positive for CD79a

Figure 6. Extranodal marginal zone B-cell lymphoma (MZBCL) with concomitant, monotypic plasmacytic differentiation. A, This extranodal MZBCL has a focal area of monocytoid differentiation (upper left) and plasmacytic differentiation (lower right corner). B through D, The plasma cells are positive for CD138 (B) and dimly positive for CD19 (C) but negative for CD20 (D). E and F, These plasma cells are positive for k (E) but negative for Lambda (F) light chains (hematoxylin-eosin, original magnificationx100 [A]; original magnification x200 [B through F]).
- can be difficult to separate from LPL. MYD88 may be helpful
Primary Cutaneous Marginal Zone Lymphoma (PCMZL)
Not in WHO book per se?
- no evidence of systemic disease by definition [19]
- thought to be derived from post-germinal center marginal zone B-cells
Present as solitary or multifocal nodules, plaques or papules usually on arms or trunk
- 7-9% of all cutaneous lymphomas, 25-57% of primary cutaneous B-cell lymphomas
- usually adults in 5-6th decade, male predominance
- rare anetodermic form, where skin is wrinkly, 2/2 loss of elastic tissue in dermis
2 subtypes: heavy chain class-switched subtype and an IgM subtype
- vast majority (70-95%) are IgG class-switched, with 40% expressing IgG4; IgA in up to 15%
- the IgM subtype is similar to MALT lymphomas at other sites
- class-switched type may be exagerrated reaction to T-cell stimulation, while IgM type likely more a true lymphoma
__________________________________________
Micro: comprised of mix of small lymphocytes, plasma cells, and lymphoplasmacytoid cells
- dense dermal peribascular and periappendageal infiltrate that spares the epidermis with a grenz zone
- may be associated with amyloid
- IgM type has predilection for subcutis, frequent follicular colonization, and has more commonly diffusely scattered rather than peripherally located plasma cells
- "lipoma-like" PCMZL assoc c HCV infx have IgM neoplastic cells and small IgM paraprotein
__________________________________________
IHC: neoplastic cells (+) CD20, CD79a, and BCL2, negative for CD5, CD10, BCL6, CD23 and cyclin D1
- IgG4-associated PCMZL not assoc with IgG4-related disease (IgG4-RD)
- CD21 shows disrupted follcilular dendritic cell meshworks
- numerous CD3+ T-cells present in most class-switched cases, potentially obscuring the neoplastic B-cells, while IgM-positive cases usually have more numerous B-cells, or equal B- and T-cells
__________________________________________
Genes: molecular studies may be useful in cases without definitive light chain restriction
- clonal IgH or IgK gene rearrangement in 92%, although benign cutaneous lymphoid proliferations show the same rearrangements in 22% [19]
- t(14;18) IGH-MALT1 in 25%, t(11;18) and t(3;14) less common
__________________________________________
DDx: Cutaneous lymphoid hyperplasia / pseudolymphomatous folliculitis
- IgG4-related disease
- other nonspecifiic reactive lymphoid infiltrates
- Secondary involvement by extracutaneous MALT lymphoma or other B-cell lymphoma
- Secondary involvement by a plasma cell neoplasm
- Primary involvement by a T-cell lymphoma (ie primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder [PC-SMTLPD])
__________________________________________
Tx: depends on extent of disease, and whether pts are symptomatic
- may be observed or treated with local excision or radiotherapy
- antiobiotic therapy may be helpful in patients with Borrelia Burgdorferi-associated PMCZL (usually in Scottish patients, not North American pts)
- antibiotics for Helicobacter pylori may be helpful in patients with positive H pylori serology, same goes for hepatitis C (HCV)
__________________________________________
Px: very indolent disease, 5-year survival ~100%
- extracuaneous spread is rare, although cutaneous recurrence is common (36-71%)
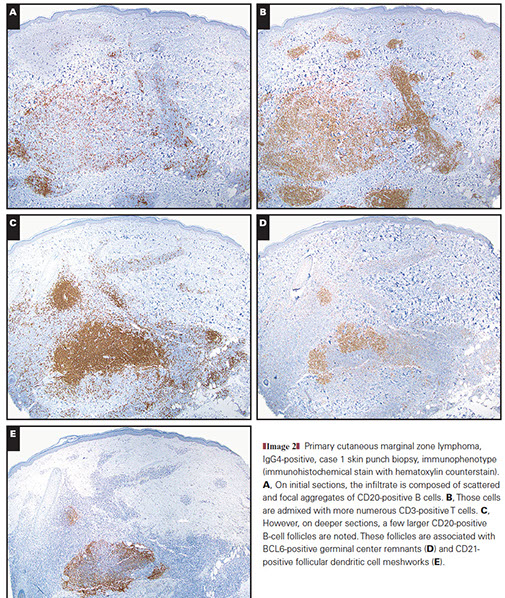
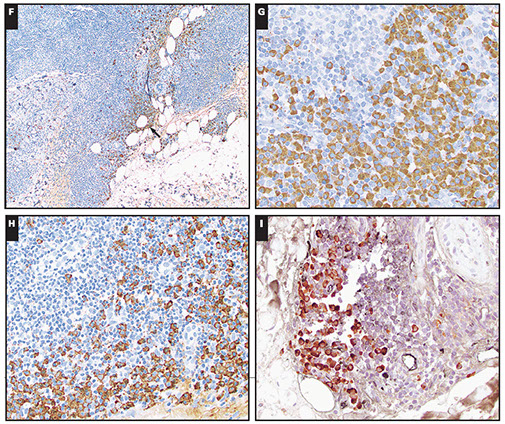
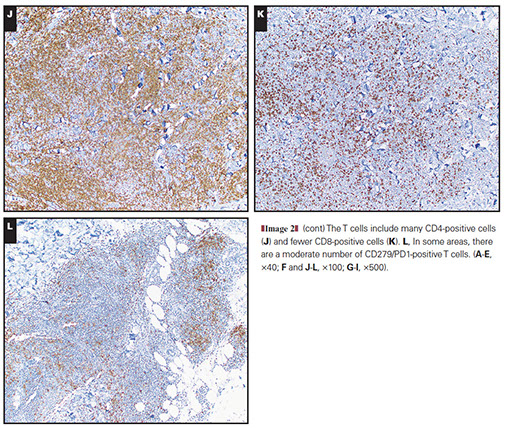

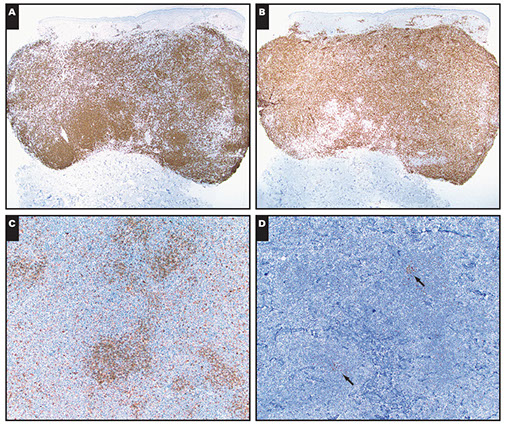
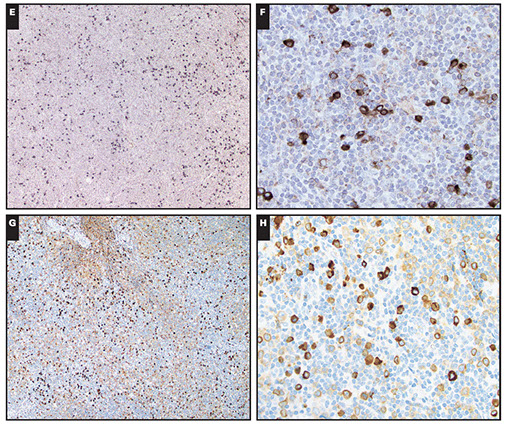







Image 1. Primary cutaneous marginal zone lymphoma, IgG4-positive, case 1 skin punch biopsy, histology (H&E). A, The biopsy shows a dense, nodular, and focally more diffuse, dermal perivascular and periappendageal infiltrate. B, The infiltrate is polymorphous with some pale-appearing areas C, It is composed of mostly small- to medium-sized lymphoid cells with round to more irregular nuclear contours that have pale cytoplasm. D, Focally, there are clusters of plasma cells (arrow), which are
noted predominantly at the periphery of the infiltrate. [19]
Image 2 (cont) F and G, IgG-positive plasma cells (arrow) are clustered at the periphery of the infiltrate. H, They include many IgG4-positive cells. I, Those cells are λ–light chain restricted (orange) with only rare κ-positive cells (black). [19]
Image 3. Primary cutaneous marginal zone lymphoma, IgM-positive, case 2 skin punch biopsy, histology (H&E). A, The biopsy shows a dense, diffuse lymphoid infiltrate that extends from the superficial to deep dermis and spares the overlying epidermis. B, The infiltrate is polymorphous with some pale-appearing areas. C and D, It is composed of many small- to medium-sized lymphoid cells with round to more irregular nuclear contours that have pale cytoplasm and are admixed with occasional larger, transformed cells. Plasma cells are scattered throughout the infiltrate rather than in clusters at the periphery. [19]
Image 4. Primary cutaneous marginal zone lymphoma, IgM-positive, case 2 skin punch biopsy, immunophenotype (immunohistochemical stain with hematoxylin counterstain). A, The infiltrate is composed of many CD20-positive B cells concentrated in vaguely nodular areas but in moderate numbers elsewhere. B, The B cells are admixed with many CD3-positive T cells. Within the nodular areas, there are a moderate number of residual IgD-positive mantle zone cells (C) and focal BCL6-
positive germinal center remnants [19]
Image 4 (cont). κ-Light-chain–restricted plasma cells are scattered throughout the infiltrate. G and H, An IgM stain highlights both the κ-restricted plasma cells and some of the neoplastic B cells. [19]
1 - 8
<
>

Nodal Marginal Zone Lymphoma (nMZL)
A rare and heterogeneous entity
~2% of lymphoid cancers, usually older adults (60 yo), M=F, usually does not involve marrow (var)
Expert pathologist agreement with consensus diagnosis in one study was 63%
- not everyone seems to be using the same criteria to make the dx of nMZL
Histopathologic issues in making this diagnosis:
- prominent monocytoid cells not always (or often not) present, complicating definitive diagnosis (in some series, the diagnosis becomes one of exclusion)
- follicular colonization seen and it may be impossible to determine if residual GC are colonized by transformed neoplastic cells or reactive. Dont overdiagnose transformation
- plasmacytic differentiation may be present (21-44%), distinciton from lymphoplasmacytic lymphoma (LPL) sometimes problematic
How many large cells are OK? [9, lecture 2]
- Variable numbers of large transformed cells described without clear-cut criteria for transformation
-- WHO doesn't accept sheets of transformed cells
- focal inc in large cells in 25%; large cells >20% of tumor cells in 57% of cases; >50% large cells in half of cases; >20% in 16% and 41-50% in 6%; "MALT+DLBCL" type
- Be careful - at least comment if there are prominent large/transformed cells
A minority of nMZL resemble LN involved by a SMZL
- Polymorphic proliferation around and infiltrating GCs without a well-defined mantle zone creating a nodular/vaguely nodular appearance - like nodes involved by SMZL
- - some cases have proliferation within an attenuated mantle zone
IHC: phenotypically not distinguishable from other marginal zone lymphomas (CD5-, CD10-, cyclinD1-) except no nuclear BCL10
- heterogeneous regarding IgD, CD43, IRF4/MUM-1, CD38
(+) Bcl-2, CD20 / 45 / 38/138 (var) / 79, PAX5, MUM1 (var), cytoplasmic Ig (var)
- neg: CD10 / 5 (+ in ~10%)/ 23 / 43 (1/2+) / 56, EBV/HIV
Newer IHC that may be helpful either in negative (vs FL -- MEF2B) or positive sense (IRTA1 in 50-73% nMZL plus MALT lymphomas)
Genes: no consistent cytogenetic abnormalities
-- do NOT have t(11;18)
- sometimes have trisomies similar to MALT lymphomas (+3, +18) or 3q27 translocation (adult and pediatric)
- <10% have the LPL-assoc MYD88 L265P mutation
Px: - considered indolent but with variable reported survivals, possibly improved in rituximab era, 72% estimated 10 year OS
- Stage III/IV: 22-90%
- pediatric cases separated differently - more often localized and have an excellent px
nMZL - scattered hyperplastic, large GC, extensive perifollicular confluent proliferation that appears as a pale area [9]
sMZL. See a GC in the bottom left, then confluent proliferation of relatively small lymphoid cells with angulated nuclei and pale cytoplasm, arrow pointing to how pale cells are infiltrating germinal center area, with a blastic element still remaining there

A minority of nMZL resemble LN involved by a SMZL - Polymorphic proliferation around and infiltrating GCs without a well-defined mantle zone creating a nodular/vaguely nodular appearance - like nodes involved by SMZL - - some cases have proliferation within an attenuated mantle zone -- left looks kinda like FL, (right) nodules look like they have a central core, like a GC, cells at periphery have pale cytoplasm and pale central nuclei.
nMZL can have a variable cytologic appearance

Higher power of above shows central nuclei with pale cytoplasm (case from above)
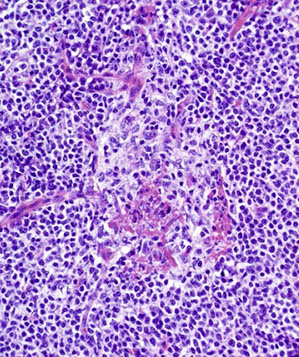
Case from above, showing GC staining as a normal GC, staining with BCL6, and negative for BCL2, but surrounded by BCL2+ IgD+ cells, which is typical of nMZL
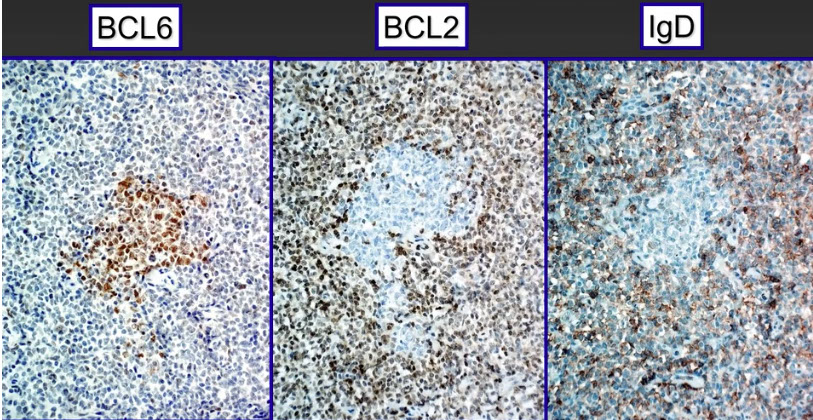

Genetics of nmzl and smzl with PTPRD, MLL2, NOTCH2, and KLF2 mutations
Patterns of PD1+ cells (A) follicular pattern with peripheral PD1 staining, 4 cases showing spectrum of PD1 expression. B. Follicular pattern with a central accumulation of intrafollicular PD1+ cells. C. Diffuse pattern with moderate extrafollicular PD1 staining. D. Normal. Strongly PD1+ cells confined to and show polarization within reactive follicles

Pediatric nodal marginal zone lymphoma (pnMZL)
- often demonstrate PTGC-like lesions (less commonly in adults)
DDx: pediatric Nodal Marginal Zone Hyperplasia (pNMZH) Pediatric nodal marginal zone B-cell lymphadenopathy of the necl: a Hemophilus influenza-driven immune disorder
- pNMZH showed a prominent nodular appearance with variable fibrosis without acute inflammation.. Within the nodules, the expanded GCs and variably sized marginal zones were colonized by activated B-cells with weak expression of IgD and lack of CD10 nd or cases test, this was confirmed by flow cytometry for surfcae Ig
- several H influenza strains are known to interact with the constant part of IgD on human B-cells leading to thier polyclonal proliferation and activation . We speculte that in vivo stimulation of IgD+ marginal zone B-cellss by this bacterium may be implicated in this particular lymphadenopathy that should be distinguished from monoclonal pNMZL
also in ddx:
- MALT lymphoma presenting in LN
- FL with marginal zone differentiation
- MZL mimic ("monocytoid-appearing" MCL, CLL/SLL with large pale-appearing proliferation centers without many paraimmunoblasts)
- Lymphoplasmacytic lymphoma (LPL, MYD88 L265P mutations)
- T-cell neoplsams
pnMZL - often demonstrate PTGC-like lesions (less commonly in adults), IgD here is highlighting mantle zone-like B-cells that are dividing up these GCs like you see with PTGC and the lymphoma is outside of the follicles
Follicular lymphoma (FL)
Neoplasm of germinal center B-cells (both centroblasts and centrocytes), usually in a nodular follicular pattern that has lost its tingle body macrophages and polarization
- if there are diffuse areas composed of >centroblasts (Grade 3), best to dx DLBCL (as the primary dx)
- germinal center ancestry of the B cells supported by mutations in Ig genes (IgVH)
*** PB-FL = Peanut Butter For Life!!! *** Paratrabecular centroBlasts Follicular Lymphoma ***
- peanut butter gets stuck to your teeth just as the lymphs in FL get stuck to the bone
Centroblasts: large (thrice the size of lymphs) transformed cells with round-oval nuclei and multiple blue peripheral nucleoli and moderate amt of cytoplasm
- used to grade FL
Centrocytes: small, monotonous cells c irregular (angled, long, twisted, cleaved) nuclei
*** C-shaped nuclei *** (mostly??)
Comprises 1/5 of all lymphomas, incidence higher in Western countries
- more likely in asymptomatic older females, in any form of lymph tissue or extranodally, that is usually widespread (involving BM in up to 70%) at time of dx
Centroblasts and centrocytes distributed randomly in nodes with effaced architecture (non-polarized)
- centrocytes in the follicle are smaller and have less irregularities than those in germinal centers
Diffuse areas = tissue lacking follicles defined by CD21/23 positivity
- dx DLBCL if Grade 3 blastic/transformed cells occupy diffuse areas
-should estimate the diffuse and follicular areas in the pato report
FL found in paratrabecular (right up next to the bone) areas in BM
Diffuse FL can be dx'd when architecture is exclusively diffuse with mostly centrocytes and few centroblasts, in addition to classic t(14:18)
Partial nodal involvement dx'd when normal areas with follicles present in same LN as FL; generally assoc c better px and low-stage dz
Micro: effaced architecture, crowded back to back follicles, monotonous cells are mostly centrocytes with invasion of centrocytes out into the interfollicular regions, dec mits, deceased centroblasts in the GC, dec macrophages, positive Bcl-2 and t(14;18), little GC morphologic variation
Beware of cases that look like they have mostly centrocytes but which are blastoid - these cases are specifically commented on in 2017 Blue Book and not considered indolent [9]
- Ki67 stains can be helpful but no agreed upon rules as to how to use it and stain isn't very reproducible (especially not useful in core needle biopsies)
-- cases with Ki67>30% in 10 randomly selected fields appeared to behave like grade 3 FL even if looked "low grade" [9]
Follicular Lymphoma Morphologic Variants
•Marginal zone differentiation (10%).
–Monocytoid cells part of the neoplastic clone.
–? Worse prognosis.
•“Floral” variant.
–Resemble progressively transformed germinal centers (PTGCs).
–More frequently CD5+
•With signet ring cells.
•Plasmacytic differentiation.
–Shows cytoplasmic light chain restriction.
Follicular Lymphoma Variants and Related Entities
•Testicular follicular lymphoma.
•In situ follicular neoplasia (formerly FL in situ).
•Duodenal-type follicular lymphoma.
•Pediatric-type follicular lymphoma
•Large B-cell lymphoma with IRF4 rearrangement
–May show follicular and/or diffuse patterns.
•Primary cutaneous follicle center lymphoma.
IHC:
(+) CD10 (increased in interfollicular areas) / 19/20/22/79a, BCL2 (in GC 9/10 grade 1-2, 50% grade 3)/6, K/L light chain restriction
-- CD10 expressed more commonly in grades 1 and 2 compared c grade 3 (though ~1/2 are + in grade 3) (possible DLBCL assoc?); CD10- cases should still be BCL6+
- MUM1 more common in grade 3, while Bcl-2 expression more freq in FL grades 1 or 2
- grade 3 more commonly expresses p53 when DLBCL component present
- new markers for germinal centers (GCs): GCETI, HGAL and LMO2, MEF2B
- neg: CD 5 / 23 / CD43-
BCL2 - don't overinterpret BCL2+ nodules that actually represent primary follicules rather than germinal centers
- not all BCL2+ neoplastic "follicles" are FL
- Some BCL2+ cases may appear BCL2- with the antibody most people use - can use "E17" antibody to pick up more cases
-CD10 seen in GC and interfollicular zones
- IRF4/MUM1 (-), can be + in some CD10- cases
- BCL2 can be absent if mutated or in high-grade FL / primary cutaneous FL / pediatric FL
-- read BCL2 cautiously bc (+) in normal germinal center (GC) and mantle zones (but these primary [resting follicles are CD10/Bcl-6 neg and dont have GC cells and are IgD-[+])??
- interfollicular cells can lose CD10 and Bcl-6
Ki-67 should be done to determine grade, in which <20% is grade 1-2, >20% grade 3
- Ki-67 very high and polarized in reactive follicular hyperplasia
Kappa and lambda stains usually not needed or helpful, but can be useful sometimes in a pinch (can usually get more kappa than lambda in GCs)
Bernard cell counting method to grade FL (usually combine grades 1 and 2) - count large transformed cells / centroblasts in 10 random but representative high-power fields - although (CAVEAT!) you must adjust for the size of your microscopes HPF!!!!
0-5 centroblasts / hpf = Grade 1
6-15 = grade 2
>15 = grade 3
-- grade 3a is when centrocytes still present, grade 3b when solid sheets of centroblasts present (much closer to DLBCL)
- Diffuse areas of grade 3 are diagnosed as DLBCL [9]
Flow: small/med sized light chain-restricted CD10+ B-cells
FISH is best cytogenetics test
- 90%+ have t(14;18)(q32;q21) which rearranges the BCL2; 20% have +7 or +18
Up to 90% of grade 1-2, <50% in grade 3 (mostly 3A).
•Secondary cytogenetic abnormalities very common (median additional chromosome breaks = 6).
•May lack BCL2 protein expression due to BCL2 mutation.
•Cannot be distinguished from IgH/MALT1 by karyotype (requires FISH).
•Negative in testicular FL, primary cutaneous FCL, pediatric type FL.
•Concurrent BCL2 and MYC rearrangements found in small minority of FL; behave similar to other FL and do not have the poor prognosis associated with HG (double hit) BCLs.
BCL2/IGH@ rearrangement in the PB of ~15% of cases (also seen c DLBCL)
3q27-28 assoc c high-grade FL
- Blood 2016: mutations in chromatin regulator/modifier genes, such as CREBBP and KMT2D (MLL2) are extremely common early events and can be potential therapeutic targets
- BCL2 rearrangements are NOT speicifc for FL, can also be seen in some DLBCL, esp of the GC phenotype, other small B-cell lymphomas (CLL or HCL), although it does exclude Burkitt lymphoma (which has the MYC rearrangement and simple karyotype)
FL without BCL2 may have BCL6 translocations
Tx/Px
Grading done by counting the # centroblasts per 10-hpf, which are involved but randomly selected
- No difference bwt grades 1 and 2; lumped together as "Grade 1-2 of 3."
- Grade 3 divided into A (with centrocytes still present) and B (sheets of blasts)
-- Grade 3a considered indolent (though not very clear) and grade 3b aggressive like DLBCL
Never correct to make dx of "follicular lymphoma, grade 3 with diffuse areas", bc would then be a DLBCL
There is a group of distinctive largely diffuse low-grade FL that can present as large localized inguinal masses
- lack BCL2 and have 1p36 deletions (often large, localized and inguinal) - 1p36 deletion is not a specific entity
Px related to extent of dz at dx
Grade 1-2 is pretty indolent; whereas Grade 3 (esp c large diffuse areas) have worse px
- multiple genetic abnormalities are assoc c poor px
Up to 35% transform into higher-grade DLBCL
Follicular Lymphoma International Prognostic Index 2 (FLIPI-2) based on rituximab tx-response groups and based on age, Hgb, size or largest node, B2 microglobulin levels, and BM involvement

FL, grade 1

FL - leukemic phase

FL Hyalinized

FL grade III

Histological appearance of nodal follicular lymphoma.
(1A) Low-power field of follicular lymphoma, Grade 1 (H&E
stain, ×40). Neoplastic follicles have vague borders and scant or absent mantle zones. (1B) High-power field of follicular lymphoma, Grade 1 (H&E stain, ×200). Tumor cells are composed of small to medium-sized cleaved cells (centrocytes) and large noncleaved cells (centroblasts).
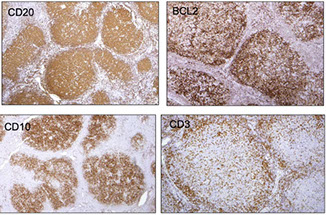
FL IHC: Neoplastic cells are positive for BCL2, and CD10. CD10 is also expressed in cells in the interfollicular region. It is important to look at the CD3 with the BCL2 because T-CELLS ARE BCL2 POSITIVE, and can be relatively abundant within active follicles [4]



Some follicular lymphomas, such as those with the floral pattern, resemble progressive transformation of germinal centers (PTGC)

Follicular lymphoma with marginal zone differentiation

FL with plasmacytic differentiation [9]
CD138
May be more related to marginal zone lymphoma


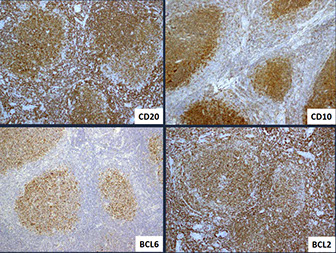
FL reporting


FL. Rare admixed centroblasts (arrow), mostly small centroblasts. The few centroblasts would argue against mantle cell lymphoma
FL Genetics

Distinguishing FL from FH [9]


Testicular Follicular Lymphoma
•Rare variant of FL.
•Reported in higher frequency in children; rare in adults.
•High cytologic grade (3A); resemble primary cutaneous FCL.
IHC: CD20+, CD10 +/-, BCL6+, BCL2-.
Genes: Similar to pediatric-type FL, absence of t(14;18) BCL2/IgH translocation.
Px: Excellent prognosis even without additional therapy.

In-situ Follicular Neoplasia (ISFN)
In-situ form of follicular lymphoma (although can be disseminated??) -- usually a situation where you are ready to diagnose follicular hyperplasia, until you get the IHC back...
Partial of total colonization of germinal centers by clonal B-cells harboring BCL2 translocation in otherwise reactive-appearing LN (or other lymphoid tissue including spleen).
•Usually incidental finding (2% random LN bxs); incidence increases with age.
•May be seen in pts with overt FL at another site, either concurrently or subsequently.
•Should be distinguished from partial involvement by FL.
- have a low rate of progression, though often assoc c prior or synchronous overt lymphomas
- should be differentiated from a partial involvement by FL, which is more likely to progress
- the degree or extent of an in situ lesion cannot be used to predict whether it will progress to overt lymphoma
Flow cytometry finds B cells c FL-like phenotype in ~1/2 LNs c ISFN
Genes: has fewer chromosomal copy-number abnormalities than focal or overt FL
- secondary genetic abnormalities present even in the earliest lesions in addition to Bcl2 rearrangements
- at low end of spectrum, cells similar to those c t(14;18)(q32;q21) IGH/BCL2 translocation can circulate in many healthy individuals
- higher levels of circulating t(14;18)+ lymphs (>10^-4 of total cells) indicates a higher risk for FL
- EZH2 mutation has also been reported (c/w early change).
Clinical significance
If no other evidence of FL on staging very low risk of subsequent FL ( 5%).
•Number of follicles involved by ISFN not predictive of subsequent risk of FL.
•High level of FL-like B-cells in PB associated with increased risk of subsequent FL.
•Additional biopsy recommended if other LAD present.
In-situ follicular neoplasia versus Partial involvement by Follicular lymphoma [9] [WHO 2017]


ISFN

Lymph node that could be taken for follicular hyperplastia, some have starry sky, some monotonous, and only some of the follicles are negative for BCL2, and others are positive.

Centrocytes are strongly positive for BCL2, and are confined to the follicle center (thety are more intensely positive than the surrounding lymphocytes
FLIS - GC with tingible-body macrophages, sinuses are intact, most have good mantle zones [9]



Duodenal-Type Follicular Lymphoma
•Rare; adults (middle-aged), M=F
•Predominantly second portion of duodenum with multiple small polyps (incidental finding); large tumors uncommon.
•Localized disease (stage IE or IIE), although not always
Low grade cytology (grade 1-2)
•CD20+, CD10+, BCL6+, BCL2+
Genes: Positive for BCL2/IgH t(14;18) rearrangement
•Similar immunophenotypic and genetic features to nodal FL, but lower frequency of other genetic aberrations than nodal FL.
•Gene expression studies and restricted IgHV usage suggest overlap with MALT, and by gene expression both show overexpression of CCL20 and MADCAM1
•Recurrent 1p36 deletion and TNFRSF14 mutations observed (similar to nodal FL).
The mutational landscape of DTFL is highly related to nodal FL, but harbors fewer multiple/biallelic mutations in KMT2D [9]
- the immune microenvironment of DTFL is distinct from nodal FL and characterized by a chronic inflammation gene signature [9]
DDx: sometimes features overlap with MALT lymphomas
Px: Pts do very well, sometimes without therapy or with just excision (no recurrences with just surgical excision, no deaths)
•Indolent disease course; excellent survival even without treatment.

Duodenal-type FL

Pediatric-type Follicular Lymphoma
Affects primarily in children and young adults (mediam age 15-18 years), M:F 10:1.
•Occasionally affect adults (therefore “pediatric-type”).
•Peripheral LAD, usually head and neck, stage I.
•Absence of BM involvement; no B symptoms.
•Excellent prognosis despite high cytology grade
LN effaced by large expansile follicles, often serpiginous.
•Follicles show starry sky pattern, attenuated or absent mantle zone, and blastoid cytology.
•Follicles composed of intermediate-sized blastoid cells (grade 3B morphology).
•Marginal zone differentiation may be seen.
•BCL2- or dim; CD10+, BCL6+, MUM1-, Ki-67 > 30% (no polarization).
•Grading not required if criteria for PTFL met
Nodal dz characterized by large expansile highly prolif follicles that usually have prominent blastoid follicular center cells rather than classic centroblasts (or centrocytes)
- BCL2 rearrangements must not be present, although there can be some BCL-2 expression
- lack BCL6 and MYC rearrangements
Similar lesions can occur in adults (though must strictly follow the criteria for dx in order to avoided undercalling a grade 3 FL)
_____________________________________
IHC: very high Ki67, BCL2 negative in GCs
_____________________________________
Genes: No BCL2, BCL6, or IRF4 translocation
- (no t(14;18) - although IGH is rearranged
•Usually lack FL-associated mutations KMT2D (MLL2), CREBBP, and EZH2.
•Deletion of 1p36 or TNFRSF14 mutations are common.
•Mutations in MAPK pathways genes (MAP2K1, IRF8) also common.
Light chain-restricted germinal centers in reactive lymphadenitis is possible! So be careful even in adults!! [9]
_____________________________________
DDx:
•Florid follicular hyperplasia (no effacement; Ki-67, K/L IHC, and/or molecular may be helpful).
•Nodal MZL with follicular colonization (IHC: CD10, BCL6, LMO2).
•Burkitt lymphoma (diffuse pattern, MYC rearrangement).
•Large B-cell lymphoma with IRF4 rearrangement, follicular pattern (MUM1+, IRF4 rearranged).
•Testicular follicular lymphoma (location, grade 3A cytology).
____________________________________
Tx: watchful waiting
_____________________________________
Px: almost all cases are local and may not need other tx other than excision
- may be considered a benign clonal proliferation with low malignant potential
Diagnostic criteria for pediatric-type FL (WHO 2017)


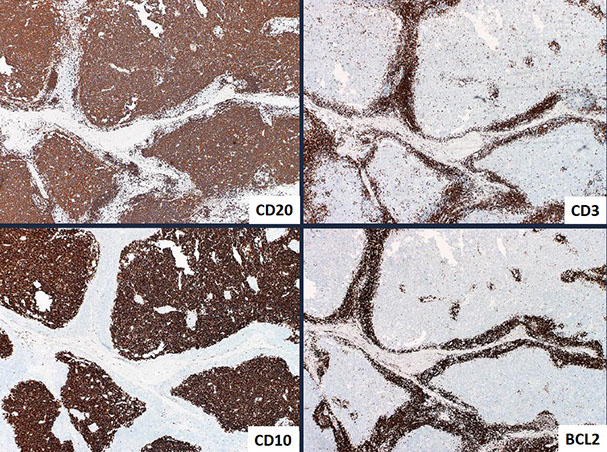
Pediatric FL


Mutational differences in conventional FL vs PTFL (boxes mean the difference is statistically significant). PTFLs generally lack mutations in KMT2D (MLL2), CREBBP, and EZH2, genes frequently mutated in usual FL... including cases negative for BCL2 rearrangements... the most common genetic aberrations are deletion at 1p36 and deletions or mutation s affecting TNFRSF14... MAP2K1 mutations are found in approximately 40-50% of cases (WHO 2017)
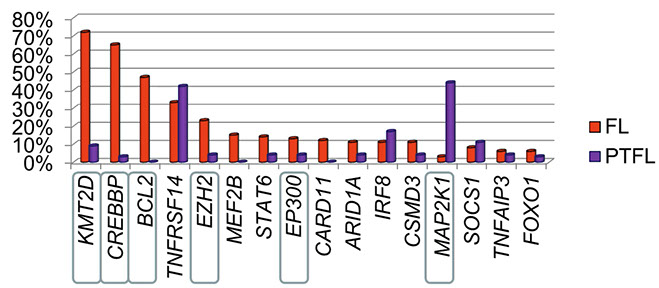
Pediatric FL. Large very expansile follicular structures, relatively monotonous. Few CD10+ cells spilling outside of follicle. In this case, IgD highlights narrow mantle zones (which is typical) also staining the germinal center areas
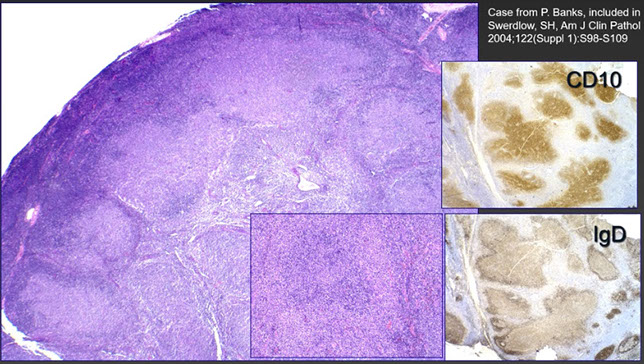

Blastoid cells in pediatric-type FL;
CB = Classic Centroblast

Large B-cell Lymphoma with IRF4 rearrangement
MC in children and young adults, in Waldeyer ring and cervical LN and are low-stage
Uncommon (0.05% of DLBCL).
•Much more common in children/young adults (but can occur in adults), M=F, low stage at presentation
•Waldeyer’s ring, head/neck LN, GI tract
•Morphology
–Follicular, follicular and diffuse, or entirely diffuse patterns
–Intermediate to large cells with open chromatin
–Follicles usually lack serpiginous configuration and starry sky pattern (unlike pediatric FL).
Micro: can have a follicular, follicular and diffuse, or pure diffuse pattern resembling FL grade 3B or a DLBCL
IHC: strong IRF4/MUM1 +, BCL6, high Ki67
- BCL2 and CD10+ in >1/2, minority are CD5+
Genes: Requires FISH studies because the IRF4 rearrangements are cryptic. Most often germinal center type
- most have Ig/IRF4 rearrangements, sometimes c BCL6 rearrangements
- uniformly lack BCL2 rearrangements
- some cases lack IRF4 rearrangement but have strong IRF4/MUM1 expression
DDx: Must distinguigh from CD10-, IRF/MUM1+ FL (which are usually assoc c DLBCL and occur in older individuals)
Px: –Favorable prognosis after treatment (chemo +/- radiation).
- More aggressive than pediatric-type FL, although pts do well c tx

(A-D) LBCL with IRF4 rearrangement. (A) Note the very large abnormal-appearing follicles in the central portion of this tonsil. (B) The neoplastic follicles have numerous transformed cells that are (C) IRF4/MUM-11 and (D) BCL6.


Blood 2016 FL updates...

Primary cutaneous follicle center lymphoma (PCFCL)
See red to purple nodules (rarely ulcerate) usually on head/neck and trunk, this is a tumor of follicle center cells such as centrocytes and centroblasts with a follicular and or diffuse growth pattern
- MCC primary cutaneous B-cell lymphoma
- rarely goes to other organs
Histo: surrounds glands and hair follicles but rarely involves the epidermis with variable mixture of follicular and diffuse growth patterns
- mostly made of medium to large centrocytes with a lesser component of centroblasts
- usually is accompanied by reactive T-cells
- also has CD21/35 + network of follicular dendritic cells, has no mantle zone
IHC: (+) CD20 / 79a / 10 (var, usual negative if has diffuse growth pattern), Bcl-6
- neg: Bcl-2, (if positive, need to r/o cutaneous involvement of systemic FL) MUM1, monotypic K/L, CD5 / 43, cytoplasmic IgM
Genes: do not, or rarely, show BCL2 rearrangements
Px: excellent, >19/20 5-year-survival
- 1/3 get cutaneous relapses, but that does not affect survival
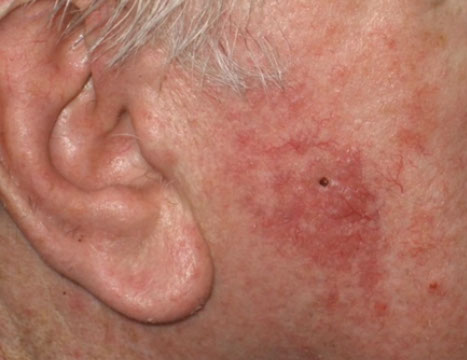




PCFCL. single papule on head
PCFCL. Diffuse and vaguely nodule, spares epidermi
PCFCL. Nodular areas with germinal centers lack macrophages and is comprised of centrocytes and centroblasts
PCFCL. CD20+ nodules
PCFCL. Ki67, not the normal staining pattern for reactive follicles
1 - 5
<
>
Mantle cell lymphoma (MCL, or MZL)
Assessment of the proliferation fraction is necessary before signing out a case of MCL!!!
- higher mitotic rates associated with worse survival
- If Ki67 >30% has worse px
- blastoid or pleomorphic have (in most studies) an adverse prognosis (may have intraobserver variability
Aggressive, incurable small B-cell cancer with diffuse / nodular prolif of monomorphic small to med centrocyte-like cells c irreg nuclear contours and CCND1 translocations
- does not have neoplastic transformed cells (centroblasts) paraimmunoblasts, or proliferations centers
-- up to 1/10 of all nHLs, in LN, BM, spleen, GI of older males, usually advanced stage at presentation (PB flow usually (+) as well)
- most cases of lymphomatous polyposis are MCLs studding the GI tract
- most have lymphadenopathy at presentation, esp involving Waldeyer ring and GI (lymphomatous polyposis)
- Classical MCL made of IGHV-unmutated or minimally mutated B cell that are SOX11+
Micro: "vaguely nodular" or diffuse in LN; sm to med size lymphs that look like centrocytes, freq mits, hyalinized vessels, monomorphic cells thickly filling in a kinda nodular mantle zone with monotonous cells (no large cancerous lymphs), histiocytes
- blastoid and pleomorphic vars are more aggressive
- small cell variant looks like CLL; marginal zone variant looks like MZL
IHC: (+) IgM/D, CD5 /19/20(bright)/CD22/43/79a, sIgM or IgD, FMC-7, Cyclin D1 (bcl-1, NUCLEAR, CCND1, PRAD1, B cells from the normal mantle zone are cyclin D1 negative), Bcl-2, sIg (bright), SOX11
- negative CD10, Bcl-6, CD23, CD99, LEF1 (although can be seen in blastoid / pleomorphic variant)
- Ki67 important in px (worse if >30%)
- in cases of MCL that are Cyclin D1 neg, will be + for SOX11, or Cyclin D2/D3 (about half of cyclin D1 negative cases have CCND mutations)
SOX11 has ~90% of cases with nuclear positivity, including Cyclin D1 negative cases
- complicated story as a prognostic indicator - better px but dismal if also with TP53 abnormalities
- MRQ-58 antibody very specific but other ab that may still be used stain many lymphoblastic lymphomas, some BL, and some DLBCL
- SOX11 normally stains scattered positive cells in GC
Flow: small FMC7+ B-cells, CD20 (bright), light chain (bright)
Genes: t(11;14)(q13;q32) bwt JH region of IGH@ and cyclinD1 (CCND1) present in almost all cases (causing cyclinD1 overexpression)
- is the primary genetic event, causing overexpression of CCND1 mRNA and protein
- cyclin D1 amplification can overcome cell-cycle suppresion of PB1, with Rb phosphorylation and E2F release (allows cells to pass from G1 to S phase)
- FISH is most sensitive assay
- WHO 2016: ATM (40-75%), CCND1 (35%), and other less freq mutations such as NOTCH1
DDx: Follicular Lymphoma (FL), Chronic lymphocytic leukemia (CLL/SLL0, Marginal zone B-cell lymphoma (MZBCL)
Px: avg survival up to 5 yrs
- high mit rate (c Ki67) assoc c worse px, may be best px indicator
- does not transform to DLBCL
Broad morphologic spectrum of MCL, the left looks almost normal. More typically you see, on the right, wide prominent mantle zones surrounding germinal centers of variable sizes.

This case of MCL has characteristic hyalinized blood vessels

This case of MCL has a more follicular growth pattern

MCL high power proliferation of relatively monotonous small lymphoid cells with angulated nuclei and dispersed chromaton, look similar to centrocytes in normal germinal centers (which is why they were originally called "centrocytic lymphoma", altugh it lacks the centroblasts of a normal gemrinal center)


MCL - proliferation of small cells that look like centrocytes [4]

Neoplastic cells in MCL positive for Cyclin D1 surrounding a germinal center [4]t
Mantle cell lymphoma (MCL)


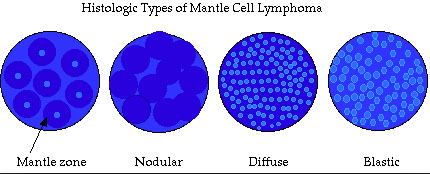

Mantle cell lymphoma (MCL) in situ and SOX11 expression. A, A case of MCL in situ in which cyclin D1+ cells are limited in the mantle zone without thickening. B and C, A Case of MCL with nuclear staining of SOX11 (panels B and C are courtesy of Yi-Hua Chen, MD, Department of Pathology, Northwestern University, Chicago, Illinois (original magnifications x40 [A] and x200 [B and C]).
Aggressive type of MCL: Blastoid (resembles lymphoblasts and have a high mitotic rate) and Pleomorphic variant (looks more like a DLBCL)

MCL FISH, BAP wiht red/green fusion on left
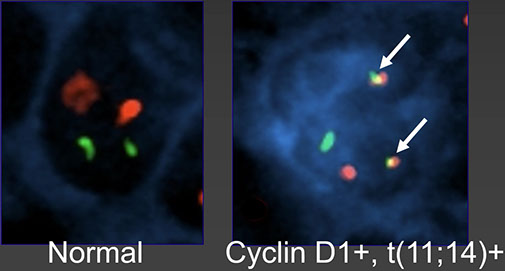

MCL - in this case neoplastic cells positive for Cyclin D1 surrounding Germinal Center, has CD5 expression that is dimmer than T-cells (seen with CD3)

Pathogenesis of classical MCL vs the blastoid variant. Blastoid variant with p53 and p15 mutations
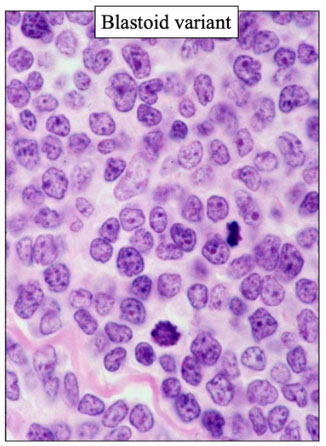
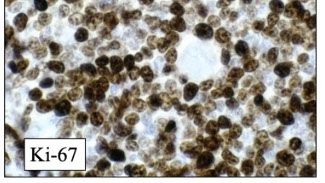
Blastoid variant of MCL with lots of mitoses, high Ki67 (right)
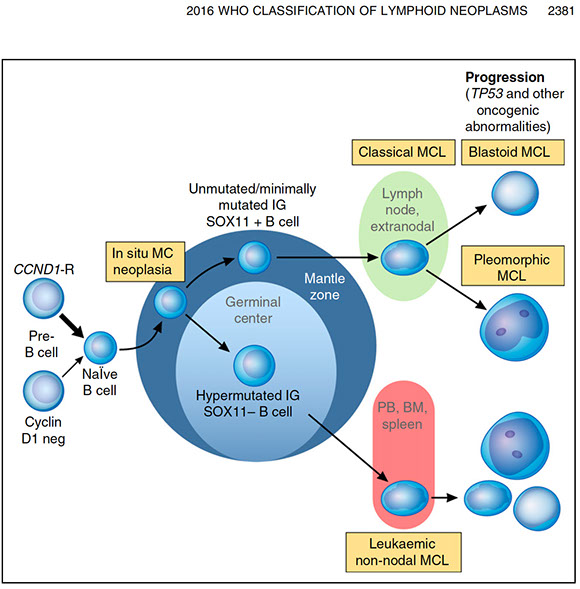
Figure 2. Proposed model of molecular pathogenesis in the development and progression of major subtypes of MCL. Precursor B cells usually with but sometimes without a CCND1 rearrangement mature to abnormal na¨ıve B cells which may initially colonize, often the inner portion of the mantle zones, representing ISMCN. These cells already have additional molecular genetic abnormalities, such as inactivating ATM mutations. They may progress to classical MCL which most frequently is SOX111, has no evidence of transit through the germinal center, and is genetically unstable acquiring additional abnormalities related to cell cycle dysregulation, the DNA damage response pathway, cell survival, and other pathways. Ultimately, progression to blastoid or pleomorphic MCL may occur. A smaller proportion of neoplastic mantle cells may undergo somatic hypermutation, presumably in germinal centers, leading to SOX112 MCL that are more genetically stable for long periods of time and which preferentially involve the PB, bone marrow (BM), and sometimes the spleen. Even these MCL, however, may undergo additional molecular/cytogenetic abnormalities, particularly TP53 abnormalities, leading to clinical and sometime morphological progression. Adapted from Jares et al31 and Swerdlow et al.2 Professional illustration by PatrickLane, ScEYEnce Studios.
Cyclin D1-Negative Mantle Cell Lymphoma
•Lack cyclin D1 and t(11;14)(q13;q32) CCND1/IgH translocation.
•Otherwise similar morphology, clinical features, and gene expression signature to cyclin D1+ MCL.
•Approximately half the cases are cyclin D2+, and have CCND2 translocations by FISH (usually Ig partner, often IgK or IgL enhancers - known as "enhancer hijacking").
- a smaller subset have upregulation of CCNE1 and CCNE2 (Blood 2019; 133(9):940-951)
•Cyclin D2 or cyclin D3 IHC not helpful (positive in other BCLs).
- infrequent cases are cyclin D1- by IHC but have CCND1 rearrangements and rare cyclin D1 cases can have cryptic CCND1 rearrangemnts
• SOX11 positive (not completely specific for BCL; also positive in some cases of BL, DLBCL, and LBL)

SOX11 useful in identifying Cyclin D1 negative cases of MCL
- SOX11 positivity is relatively specific, can be positive in Burkitt lymphoma (25%) and in hairy cell leukemia (50%)
SOX11-Negative Mantle Cell Lymphoma
Ribera-Cortada I et al., Mod Pathol 2015;28:1435-47.
•SOX11:
–Positive in 90% of MCL (including cyclin D1- cases and blastoid/pleomorphic variant).
–May block B-cell development.
–Blocking SOX11 may promote plasmacytic differentiation.
•SOX11-negative MCL:
–More often non-nodal, small cell variant, plasmacytic differentiation, mutated IgHV, better survival.
–Less often male, CD5+, require chemotherapy.

Unusual case of CD10+ mantle cell lymphoma (MCL). A, The MCL shows mantle zone and diffuse growth pattern. B and C, The MCL cells are positive for BCL1 (B) and weakly and partially positive for CD10 (C) (CD10, original magnification x100 [A and C]; original magnification x100 [B]).
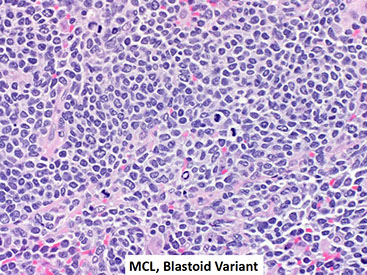
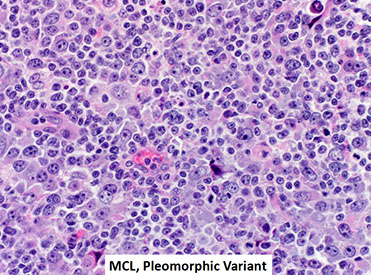

from Blood 2016 updates

Leukemic non-nodal mantle cell lymphoma
- Leukemic nonnodal MCL has IGHV-mutated SOX11- B cells that usually involves the PB, nodes, marrow and spleen
- pts present with Splenomegaly, BM and PB involvement
- usually indolent, though can have additional TP53 mutation leading to aggressive dz
- SOX-11 usually positive when BCL1 negative
In situ Mantle Cell Neoplasia (ISMCN)
- previously Mantle Cell Lymphoma In Situ (MCLIS)
Clonal CCND1+ B cells grow in the mantle of LNs that look reactive, but do not cause expansion of the mantle zone of the LN, and they don't invade into the follicles [5]
- unknown if this is a precursor to MCL
- must distinguish from overt MCL with a mantle zone growth pattern
- should still have the translocation
Presence of cyclin D1+ cells restricted to the mantle zones (not expanded), in otherwise hyperplastic-appearing lymphoid tissue with preserved architecture.
•SOX11+ (44%): M > F, CD5+ > CD5-; several pts with progression.
•SOX11- (56%): F > M, CD5- > CD5; no progression.
•Must be distinguished from MCL with mantle zone pattern (architectural distortion, expanded mantle zones)
In situ mantle cell neoplasia
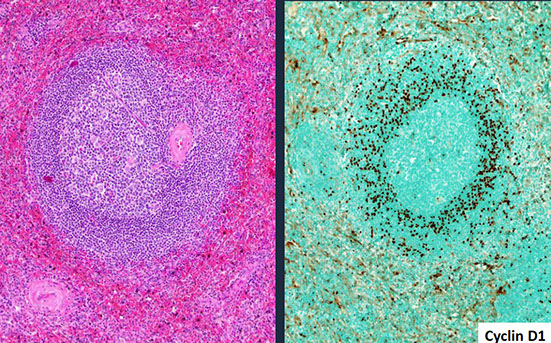
ISMCN, characteristic Cyclin D1 staining
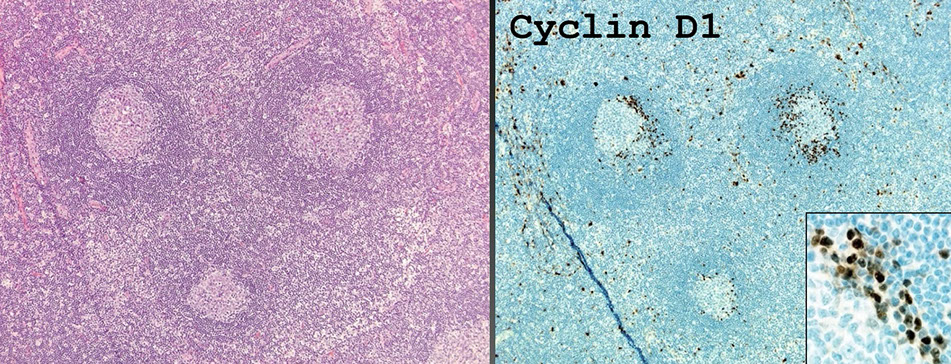
Diffuse large B-cell lymphoma (DLBCL), NOS
B- cells have nuclei larger than macrophages (>2x normal small lymphocyte); is a large B cell lymphoma that does not conform to other lymphoproliferative neoplasms
- ~1/3 nHLs or more! (MCC non-Hodgkin lymphoma) usually in the elders (60s); can present as rapidly enlarging LN
- usually de novo, but can evolve from other ca's
- ~1/2 are extranodal at dx, usually in GI (esp stomach and ileocecal valve), but can be anywhere, tumors grow fast
~1/10 c BM involvement at dx (not very common)
•Medium to large B-cells in diffuse growth pattern
NOS = Does not fit into a specific subtype large B-cell lymphoma
____________________________________
Micro: LN architecture partially / diffusely effaced by large lymphs, usually in at least interfollicular area
Several variants (questionable prognostic significance; not necessary to specify in dx):
- Centroblastic variant - pale blue cytoplasm c vesicuar chromatin (2/2 margination), multiple nucleoli [usually close to membrane] (2-4 nuclear bound nucleoli)
- Immunoblastic variant - amphophilic cytoplasm, eccentric nuclei c a single central nucleolus (>90% cells with single central prominent nucleoli)
- Anaplastic variant - like ALCL or metastatic carcinoma (bizarre pleomorphic large cells)
____________________________________
IHC: (+) B-cell markers (CD19/20/22), CD45, bcl-2, Ig Markers (but not plasma cell markers), CD30 (esp anaplastic variant), variable CD5 (+ in 10%) /10, bcl-6
- up to 70% of new DLBCL NOS is CD5 and CD10 neg
- can use combo of CD10, Bcl6 and MUM1 to differentiate GC and non-GC variants (GC has better px)
- get a CyclinD1 if CD5+ to r/o blastoid MCL
- high Ki67 (usually in 2/3 up to 99% of cells)
____________________________________
Cytogenetics
•Absence of MYC rearrangement and BCL2 and/or BCL6 rearrangement
Diffuse large B-cell lymphoma, NOS What other testing is required?
•Cell of origin (COO) subtyping
–Germinal center B-cell phenotype (GCB)
–Activated B-cell phenotype (ABC)
–Unclassified
•Why?
–Required for enrollment in most clinical trials
–Bortezomib, lenalidomide, ibrutinib may preferentially benefit DLBCL patients of ABC type
Additional recommended testing: “double-expresser status”
•Definition: DLBCL, NOS that coexpresses MYC and BCL2
•Recommended cut-offs:
–BCL2 ≥ 50%; MYC ≥ 40%
•~1/3 of all DLBCL, NOS
•MYC IHC not a surrogate for MYC FISH!
–Does not correlate with status of MYC rearrangement (mutation, amplification), FISH is the gold standard
Double expressers
•More common in ABC subtype (~50% of ABC, ~15% of GCB)
•2/3 DE are ABC type
•Difference in outcome in ABC vs GCB may be attributable to MYC/BCL2 coexpression
____________________________________
Double / triple hit lymphoma - clinical features- MYC, BCL2 and/or BCL6 rearrangements
-Middle aged adults or older adults
--- Not typically associated with immune suppression, if present in rare cases most likely coincidental
- Some patients have history of follicular lymphoma
MYC rearrangement represents transformation event in pre-existing low-grade FL with t(14;18)/IGH-BCL2
- Widespread disease, often with involvement of bone marrow, CNS or other extranodal sites
- Markedly elevated serum LDH, often >3x upper limit of normal
- Tx: not well established
--- Large multi-center birth try specked it steady suggested some benefit of intensive therapies (R-EPOCH, R-hyper-CVAD) over RCHOP but high rates of early treatment failure and death Potential but limited role for auto-SCT and relapsed/refractory disease
Pathologic spectrum
Some with monomorphous cell population and conspicuous starry sky pattern closely resembling Burkitt lymphoma
- Others with morphologic overlap with BL, but greater variability in nuclear morphology or blastoid morphology resembling lymphoblasts
- Many cases in the indistinguishable from DLBCL-NOS
Immunophenotype
- GCB cell-of-origin (COO): CD 10/BCL6 typically positive
- MYC overexpression by IHC (usually, not always)
- BCL2 usually positive (particularly MYC/BCL2 DHL)
- Ki-67 proliferation index typically high, but often less than 95%
Genetics
Complex karyotype with many numerical and structural aberrations
- MYC partner: may be IG gene but non-IG gene more likely than in BL
- EBV negative by RNA in situ hybridization (EBER)
____________________________________
Double-Expressor Lymphoma (DEL)
-Utility of MYC immunohistochemistry
--- Prognostic marker with PCL2HC
- DLBCL with concurrent high expression of MYC ≥40% and BCL2 ≥50%
--- 20 to 30% of DLBCL (vs DHL 5 to 10% of DLBCL)
- Worse prognosis than non-DEL, though better than double-hit lymphoma
- No distinguishing morphology
- Most are ABC/non-GCB subtype by immunohistochemistry
____________________________________
Other things to consider
• CD5
–de novo CD5+ DLBCL may have poorer prognosis
–Relatively rare in the Western world (3-6%), more common in Asia
–A/w BM involvement, CNS relapse, ABC subtype, worse px
- may be associated with intravascular or sinusoidal disease
- need to have Cyclin D1 negative to distinguish from pleomorphic mantle cell lymphoma
- often suspected or detected by flow cytometry
–No clear therapeutic implications
–Important to exclude pleomorphic mantle cell lymphoma or Richter transformation of CLL.
____________________________________
•CD30
–Outcome data inconsistent
–Anti-CD30 therapy may be considered
____________________________________
Genes:
3 distinct subtypes based on cell-of-origin (COO) [11]:
1. Germinal center B-cell like (GCB)
2. Activated B-cell like (ABC)
3. Primary mediastinal (PMBL) - although this is considered in its own diagnostic category
- GCB and ABC type each comprise ~50% of DLBCL NOS
- significantly better outcome in GCB group (pre-rituximab era)
- somatic hypermutations in variable regions of Igs; ABC vs GCB differ in terms of chromosomal alterations, activation of signaling pathways, and clinical outcome;
-- the molecular categories and the immunophenotype do not always match up
- 1/3 have 3q27 region involving BCL6 gene, specifically in the Activated B-cell-like (ABC) phenotype
- 1/4 have t (14;18) of BCL2 gene [assoc c follicular lymphoma] esp the Germinal Center B cell-like (GCB) phenotype
- MYC rearrangements rare in ABC type, (even if MYC+ by IHC) seen in 5-15% (60% of those c an IG gene / 40% non-IG gene) and is freq assoc c BCL2, or less commonly with BCL6 in double- or triple-hit lymphomas (now called High-grade B cell lymphoma (HGBL) with rearrangements of MYC and BCL2 and/or BCL6
-- MYC protein expression in higher proportion of DLBCL (30-50%) and is assoc c concomitant expression of BCL2 (20-35%) (double-hit)
MYD88 mutations in DLBCL correlate with extranodal locations and ABC subtype
- certain subtypes, such as those arising in immune-privileged sites of the testes and CNS, are particularly likely to have MYD88 mutations (up to 70%)
- in some subgroups they impart a worse px, however, this has not arisen as a clinically significant independent progostic marker in DLBCL
- testing for MYD88 is not routinely done in DLBCL
____________________________________
Px: poor if older age (>60 yo), poor performance status, advanced Ann Arbor stage (3-4), extranodal involvement >2 sites, high serum LDH
- GCB-like DLBCL has better response to tx
Diffuse large B-cell lymphoma, NOS Morphology

Centroblatic and Immunoblastic morphology of DLBCL, can have a mix of both

DLBCL plasmablastic morphology

DLBCL workup

Diffuse large B-cell lymphoma, NOS Double expresser survival


Hans method for stepwise eval of DLBCL from bloodjournal.org. Need >30% cells positive to call it positive
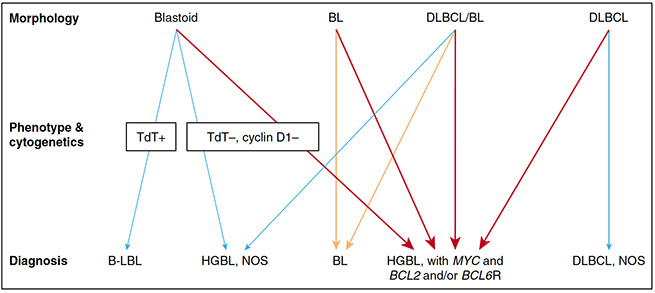
Figure 4. Diagnostic approach to HBCLs. Lymphomas that potentially fall into the HGBL categories can morphologically resemble B-lymphoblastic leukemia/lymphoma (B-LBL), BL, and DLBCL as well as lymphomas that are intermediate between DLBCL and BL (DLBCL/BL). These distinctions can be very subjective. The orange arrows indicate cases with a BL phenotype and a MYC rearrangement without BCL2 or BCL6 rearrangements (“single hit”). The red arrows indicate cases with MYC and BCL2 and/or BCL6 rearrangements (“double or triple hit”). Neither MCLs, subtypes of LBCLs, nor Burkitt-like lymphoma with 11q aberration are indicated in this diagram. Adapted from Kluin et al89 with permission. Professional illustration by Patrick Lane, ScEYEnce Studios.


Diffuse large B-cell lymphoma, NOS Double expresser (MYC/BCL2) assoc c worse survival and inc CNS relapse

Double-hit lymphoma (MYC/BCL2) patient survival vs Burkitt lymphoma and IPI-matched DLBCL


Survival of double-expressor lymphoma (DEL) vs DHL
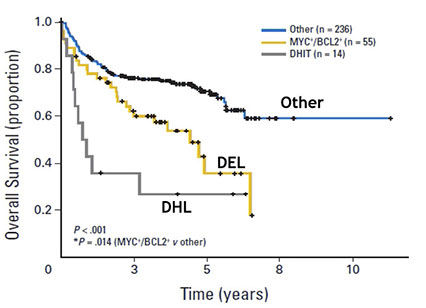
CD5+ de novo diffuse large B-cell lymphoma, not otherwise specified with nongerminal center phenotype. A, Centroblast-like, atypical lymphoid cells with frequent mitosis and scattered apoptotic bodies. B through E, Atypical lymphoid cells are positive for CD20 (B) and negative for CD3 (C) but have an aberrant expression of CD5 (D) and BCL2 (E). F through H, The atypical cells are positive for BCL6 (F) and MUM1 (G) but negative for CD10 (H) and BCL1 (data not shown) (hematoxylin-eosin, original magnification3200 [A]; original magnification3200 [B through H])

T-cell/histiocyte rich large B-cell lymphoma (TCHRBCL)
Characterized by a limited number of scattered large B-cells embedded in a background of abundant T-cells and histiocytes
- may arise de novo or progress from NLPHL
- typically presents in middle aged men with systemic involvement including fever, malaise, splenomegaly and /or hepatomegaly
- frequently involves the BM, spleen and liver
- >50% advanced stage (med-high risk) at presentation
- aggressive clinical course, often refractory to chemoherapy
- IPI highly correlated with prognosis
- multidisciplinary approach and staging essential for diagnosis and distinction from NLPHL, especially variant patterns
Micro: scattered big B cells in a background of diffuse prolif of small lymphs (mix of CD4/8+) and histiocytes
- seen in paratrabecular lymph aggs
- may appear to contain centroblasts, immunoblasts, L&H cells, RS cells
IHC: (+) (similar to DLBCL) pan B markers and bcl-6 (large B cells),
- negative CD15/30 B cells, CD57+ T cells, T cell rosettes, small B lymphs, absent CD21/23+ FDC meshwork, EBV-
- patients with lack of FDC meshwork have higher stage at presentation, no B syptoms at diagnosis, were associated with frequent relapses and required chemo
DDx: NLPHL (has FDC networks)
Approach to diagnosis in the NLPHL/THRLBCL spectrum
- mixed nodular and diffuse nodal architecture
-- Look for the presence of atypical cells and the company they keep
-- Look for an aggressive or disease component
- Look for a low grade component
Immunophenotyping -- First tier: CD20, CD3, CD30, CD15, PAX5,
- FDC markers (CD 21), : Useful to detect the presence and extent of nodularity
- Additional: EBV/Eber, CD45, OCT2, BOB1
- Use caution in the interpretation of PD1 (can be seen in reactive conditions, PTGC)
- Flow cytometry and molecular studies may be negative and not as helpful to make the dx
- WHO 2016 recommended terminology: "LPHL, THRLBCL-like
Px: usually late stage, poor px
CD20+ TCRBCL, although there is an almost total lack of smaller CD20+ B-cells in the bacground

T-cell/histiocyte-rich large B-cell lymphoma (clockwise from upper left): H&E, CD20, BCL6, CD3, CD68)

DDx: NLPHL vs THRLBCL

Primary DLBCL of the CNS
avg 60 yo at dx, see supratentorial mass that mimics GBM on imaging, possible presenting as recurrence of an intraocular lymphoma
Micro: cancerous B cells in perivascular cuffs
IHC: (+) pan-B cell ags
Genes: overexpression of bcl-6 c BCL6 rearrangement
- BCL2 rearrangement is rare
Primary cutaneous DLBCL, leg type
Rare, in elderly women
Micro: immunoblastic, round cell morphology
IHC: (+) Bcl2 (unlike PC follicular lymphoma), MUM1, FoxP1, Bcl6
- neg: CD10, no CD21 meshwork
Genes: MYD88 mutation associated with worse prognosis
Tx: combination chemotx
Px: poor
EBV+ DLBCL, NOS
previously EBV+ DLBCL of the elderly (but can occur in wide age range)
Rare; mostly in elderly Asian old people; EBV+, though now has been increasingly seen in youger pts
•<5% in Western world; 5-15% among Asian and Latin American populations.
•Most pts > 50 y/o, but does occur in younger pts
•ABC > GCB
MC extranodal sites are lungs and GI tract, though variable clinical presentation
NOS = Does not fit into other EBV+ B-cell neoplasms; need to rule out:
•Plasmablastic lymphoma, which frequently have IGH/MYC translocations (or variants)
•Lymphomatoid granulomatosis
- activated B-cell type of DLBCL (have mutations in CD79B, CARD11, and MYD88)
•DLBCL associated with chronic inflammation
•EBV positive mucocutaneous ulcer
•Post-transplant or other immunodeficiency associated lymphoproliferative disorders
- can also look like classic Hodgkin lymphoma, or even T-cell rich large B-cell lymphoma
Micro: big overlap between this and other EBV-related lymphoid proliferations, even EBV+ classic Hodgkin lymphoma (EBV DLBCL NOS also has RS-like cells)
IHC: positive for pan-B cell antigens (CD19, CD20, CD22, CD79a, PAX5) and has an activated B-cell immunophenotype (positive for IRF4/MUM1 and negative for CD10 and usually BCL6)
- CD30 frequently positive, CD15 sometime expressed
- LMP+ in >90% of cases
- usually PDL1 and PDL2 positive
- usually more than 80% of the tumor cells are EBV+
Genes: clonality of IG genes and EBV can be done molecularly, and is useful to distinguish from reactive hyperplasia and infectious mono
- some cases can have restricted/clonal T-cell receptor responses
- IG translocations are uncommon
- mutations in
Px: poor in older pts, good in younger pts
Chi Young Ok et al. Clin Cancer Res 2014;20:2338-2349; Hong, et al Ann Oncol 2015; 26:548–555 Nicolae, et al; Blood. 2015;126(7):863-872
- cases with a T-cell/histiocyte-rich large B-cell lymphoma-like or polymorphic pattern seem to do better than monomorphic EBV+ DLBCL in younger pts
- Positivity for CD30 and EBNA2 may indicate a worse px
EBV DLBCL NOS

EBV+ Large B-cell lymphoma, a true biologic spectrum [23]


Fig.13.117 EBV-positive diffuse large B-cell lymphoma. A This polymorphic lesion shows geographical
necrosis. B There is a mixed proliferation of immunoblasts and medium-sized lymphoid cells, as well as small reactive lymphocytes. C Monotonous proliferation of immunoblast-like or Hodgkin/Reed-Sternberg-like cells with prominent central nucleoli. [7]
Fig.13.118 EBV-positive diffuse large B-cell lymphoma with a T-cell/histiocyte-rich large B-cell lymphoma-like pattern. A Scattered large tumour cells are observed in a
lymphohistiocytic microenvironment. B Scattered large tumour cells are positive for CD20. C In situ hybridization for EBV-encoded small RNA (EBER) highlights scattered
tumour cells. [7]
EBV+ Mucocutaneous ulcer
Different from EBV+ DLBCL bc of self-limited growth potential and response to conservative management
- mucosal or skin ulcers are usually shallow and sharply circumscribed and can appear with advanced age and/or immunosuppression
Micro: atypical lymphs are large and pleomorphic immunoblasts c frequent Hodgkin and Reed-Sternberg (HRS) morphology
- prominent rim of T-cells at lesion base
- has underlying polymorphous infiltration of small lymphs, immnoblasts, and atypical larger lymphs with mix of plasma cells eos and histiocytes
- variable angioinvasion and tissue necrosis
IHC: Immunoblasts have (+) CD30, MUM1, PAX5, OCT2, variable CD20/45/15/79a, BCL6
- EBER positive with LMP1 colocalization (suggesting type II latency) in infiltrating small B cells, plasmacytoid apoptotic cells and immunoblasts
- ~40% have monoclonal Ig rearrangement, indicating clonal EBV-driven B-cell proliferation
- monoclonal or clonally restricted T cells in ~70% of cases
Tx: many respond with reduction of immunosuppression alone
Px: generally thought to be b9, but can be progressive and debilitating
EBV+ MCU [23]

Another case of EBV+-MCU [23]

DLBCL associated with chronic inflammation
( same as EBV+ Large B-cell "Lymphoma" Associated with Chronic Inflammation ?? [23] )
Rare; EBV+; Classically arises in chronic pyothorax
- typically occur in immunocompetent patients
- a subset arise in fibrin thrombi or amaorphous material adjacnent to cavities, prosthesis, grafts, myxoma
Px: clinically indolent, seldom disseminate
- localized alteration in host immune surveillance?
- similarity with breast implant-associated ALCL
- differ from from pyothorax-associated lymphoma as they are non-invasive and do not form mass lesions
- using :arge B-cell lymphoam" name may cause overtreatment
EBV+ large B-cell "Lymphoma" associated with chronic inflammation [23]


Fibrin-associated diffuse large B-cell lymphoma
- Unusual form of DLBCL associated with chronic inflammation, is not mass-forming and does not directly produce symptoms
- discovered incidentallyon histological examination for reasons other than lymphoma
- has been suggested to call this group fibrin-associated EBV+ large B-cell lymphoma
Micro: specimens have fibrinous materials, as in the walls of pseudocysts, lesions or matierials in the cardiovascular system, wear debris, and chronic subdural hematomas
- single and small aggregates of large lymphoma cells in only small foci in fibrinous or amorphous material
- lymphoma cells with irregular nuclear foldings, coarse chromatin, distinct nucleoli, and amorphous cytoplasm
- mitoses easily identified, and admixed apoptotic bodies prominent
- chronic inflammatory cell infiltration in the background or vicinity is NOT prominent
IHC: similar to pyothorax-associated lymphoma with expression of B-cell lineage markers and an activated B-cell phenotype
- EBV usually +, with type III latency (typically EBNA2-positive)
Px: unlike pyothorax-associated lymphoma, outcome is highly favorable, even in surgical excision alone
- one report (however) raised the possibility of progression to an infiltrative tumor, an incidental DLBCL associated with chronic inflammation in a chronic subdural hematoma accompanied by brain parenchymal infiltration
Lymphomatoid granulomatosis (LYG)
Rare, EBV-(+) B-cells with lots of reactive T-cell infiltrate and plasma cells / histios in an angiocentric/-destructive process resembling vasculitis in bilateral lower lungs of Western adult males; usually extranodal in lung, skin, and brain
- immunodeficiency is a risk factor
- present with lung sx (cough, dyspnea) and cancer sx
- lung nodules vary in size
Micro: lots of T-cells with some EBV-(+) B cells
- only skin shows predominant granulomatous rxn
- prominent vascular changes c invasion of vascular wall
- granulomas uncommon
IHC: (+) CD20
- negative CD15
Genes: can see some Ig gene clonality in higher grade dz
Grading: done by looking at proportion of EBV-(+) B-cells; most important to ddx grade 1/2 from 3
- Grade 3 have pretty occasional large atypical EBV-(+) B cells identified by CD20, lots of EBV-(+) B-lymphocytes (>50/hpf) and also has lots of necrosis and occasional Hodgkin-like cells
- if there are way too many large atypical cells, may dx as DLBCL
– Grade 1 <5 EBER+ per hpf
– Grade 2 5-20 EBER+ per hpf
– Grade 3 >50 EBER+ per hpf
Px: usually aggressive
Lymphomatoid granulomatosis (LYG)
http://pathology-home.com/new/2008052701.asp

Primary mediastinal (thymic) large B-cell lymphoma (PMBL)
~3% of NHL, is a subtype of DLBCL, but different in clinical, pathologic and genetic features
MC in young adult (35 yo) females (2:1 F:M) that probably originates in the thymus, found in anterior-superior mediastinum [1] rapidly progresses
- BM and non-mediastinal LN involvement uncommon
- called a "gray zone lymphoma (aka B-cell lymphoma, unclassifiable, with features between diffuse large B-cell lymphoma and classical Hodgkin lymphoma)" if has overlapping features with nodular sclerosis classic Hodgkin lymphoma (cHL-NS)
____________________________________
Sx: pt present with large anterior mediastinal mass that directly invades nearby organs
- may have vena cava syndrome, B symptoms, or airway obstruction
____________________________________
Micro: large cells c lots of pale cytoplasm and circular nuclei, although the nuclei may be somewhat irregular with prominent red nucleoli resemble RS cells of HL; or have lots of cell cytoplasm like lacunar cells in cHL-NS
- sclerosis c B cells trapped by collagen fibrosis (compartmentalizing alveolar fibrosis)
- may see thymic stuff (Hassals corpuscles)
Dx: Must exclude BM or LN as a primary systemic cause to dx
- thus, BM should be negative for malignancy
____________________________________
IHC: (+) CD19,20,22,45, 79a, 30 (weak, in 80%), IRF4/MUM1 (75%), CD23 (70%), BCL2/6 (less common, ~50%), MAL antigen, CD54, 95, germinal center markers (b/c thymic B cell origin; CD10/23, BCL6), and B-cell transcription factors (BOB.1, OCT-2 and PU.1), PAX-5
- negative: sIg, MHC I n II, CD5/10/15
- Ki-67 varies bwt 10-67%
Typically are of non-germinal center B-phenotype (CD10-, BCL6+ MUM1+)
- CD23 relatively frequent
Flow cytommetry: abnormal B-cells c above stains, although the surface Igs are messed up and so cannot be evaluated using K/L
____________________________________
Genes: Altered MAL gene c gains in 9p24 (75%, JAK/STAT pathway, prolongs half-life), 2p15 (50%; REL protooncogene encoding NF-kB [2]); Xp11.4-21; Xq24-26 [1] - Xq gains appear with 9p gain (?) [3]
- commonly find REL accumulating in nucleus
- BCL2/6 and MYC rearrangements absent / rare
- amplification of REL and BCL11A at 2p [1]
- inc exp of JAK2, PDL1, PDL2 at 9p [1]
- MAL protein expression (in 70%) believed to be 2/2 thymic B-cell origin [2]
- FIG1 also expressed in DLBCL
-PTP1 gene mutations also in NSHL [4]
- Loss of MHC
Transcriptional profile shares many features with classic Hodgkin Lymphoma (cHL)
- JAK-STAT pathway activation with PDL1/PDL2 updregulation
- activation of NFKB survival pathway
- recurrent fusions involving CIITA
- somatic mutaions involving PTPN1
---- PMBL and cHL may arise from a common precursor medullary thymic B-cell that secondarily acquires distinct genetic changes
____________________________________
Tx: EPOCH-R has promise to eliminate need for chest radiation[4]
- bc most pts are young females, rad tx assoc c inc risk breast ca
____________________________________
Px: good response to chemo
- poor px with more invasion; only rarely involves BM, even in recurrence [1]; px is relatively better than DLBCL
- PMBL molecular signature assoc c good px
- 40-80% complete remission; 50-80% 5-year survival 50-80%
Tend to involve unusual extranodal sites at relapse (kidney, adrenal, liver, ovary, CNS)
PMBL. Left shows fairly diffuse architecture, right is CNS relapse with thin bands of fibrosis, compartamentalizing the cells into packets (packeting-fibrosis), may see multilobulated nuclei and clear cytoplasm, can appear Reed-Sternberb (RS)-like (maybe becaues of shared genetic characteristics with cHL?)



PMBL

Intravascular large B-cell lymphoma
Aka nagioendotheliomatosis, angiotophic lymphoma, or intravascular lymphomatosis
- sx caused by large B-cells obstructing vessels
- rarely involves LNs
ALK positive large B-cell lymphoma
Aggressive B-cell neoplasm
Nodal involvement most common with diffuse or sinusoidal growth
Plasmablastic or immunoblastic morphology
_____________________________________
IHC
Strongly positivity for ALK, usually with granular cytoplasmic staining
- Positive for plasma cell markers (MUM1, CD38, CD138, EMA) with cytoplasmic heavy chain usually IgA and light chain restriction
- Mostly negative for CD45, CD30 and pan B-cell antigens (CD20, CD79a, PAX5)
- Aberrant expression of T-cell cytotoxic markers
- Cytokeratin positivity recorded in occasional cases
_____________________________________
Genes
Underling ALK rearrangement typically (CLTC-ALK fusion)
No association with EBV (EBER) or HHV8
ALK+ LBCL. (clockwise from upper left) H&E shows immunoblastic morphology with abundant pinkish cytoplasm (indicative of plasmablastic differentiation), ALK1+, CD30+ (although usually are not CD30+), IgA+, Kappa-restricted, and aberrant expression of perforin+ and` CD2+ [11]

Plasmablastic lymphoma (PBL)
Rare; is a prolif of large cells most similar to B immunoblasts / plasmablasts that have IHC similar to plasma cells and are CD20 negative
- first described in mouth; assoc c HIV+ and other immunodeficient pts (post-transplant, elderly); M>F, avg 50's-60s; usually in extranodal sites of head and neck (oral cavity>GI tract>others)
- most patients have EBV+ cancer cells
- will be HHV8+ if assoc c Multicentric castleman's
- in BM in 75% of HIV+ pts, 50% post-transplant, 25% without immunodeficiency
- some pts may have a paraprotein
- may be the result of a plasmablastic transformation of myeloma
____________________________________
Histo: spectrum of cells with big immunoblastic features to looking more like plasma cells;
- see lots o mits
- may have apoptotic bodies and tingible body macrophages
- monomorphic plasmablastic morphology usually seen in cases from oral cavity of HIV+ pts
- plasmacytic differentiation seen more in extranodal sites and LNs (should consider an anaplastic or plasmablastic plasma cell myeloma)
Postulated normal counterpart is a plasmablst (a blastic proliferating B cell that has switched it phenotype to the plasma cell gene expression program)
____________________________________
IHC: (+) CD79 (var, 40%), CD38 / 138 / 30, IRF4/MUM1, PRDM1 (aka BLIMP1), XBP1, cytoplasmic immunoglobulins (var), EBV (by EBER ISH, in 60-75%), Ki67>90%, EMA and CD30 frequently expressed, sometimes CD43 and CD45RO (T-cell markers)
- neg / weakly positive: CD20 / 45 / 43 / 56 (in 25%), PAX5, Cyclin D1, BCL2, BCL6, infiltrating T-cells are usually sparse, LMP1 expression rare, HHV8 negative
____________________________________
Genes: clonal IGH expression seen. even when immunoglobulin expression not detectable
- frequently have complex karyotypes
- MYC translocation in 1/2, usually in EBV+ cases
____________________________________
DDx: LBCL, they look very similar esp in the oral cavity (the oral mucosa type of PBL)
-- some subtypes of LBCLs have plasmablastic phenotype (ALK-positive LBCL and HHV8-associated lymphoproliferative disorder) and may be a transformation of a small B-cell lymphoid neoplasm (though these cases do not have immunodeficient pts and EBV is negative and MYC translocation rarely seen)
-- - plasmacytic differentiation seen more in extranodal sites and LNs (should consider an anaplastic or plasmablastic plasma cell myeloma)
-- history of immunodeficiency and EBV+ cells useful in establishing dx of PBL
(if distinction not possible, its ok to say "Plasmablastic neoplasm, consistent with PBL or anaplastic plasmacytoma")
____________________________________
Px: poor (very aggressive)

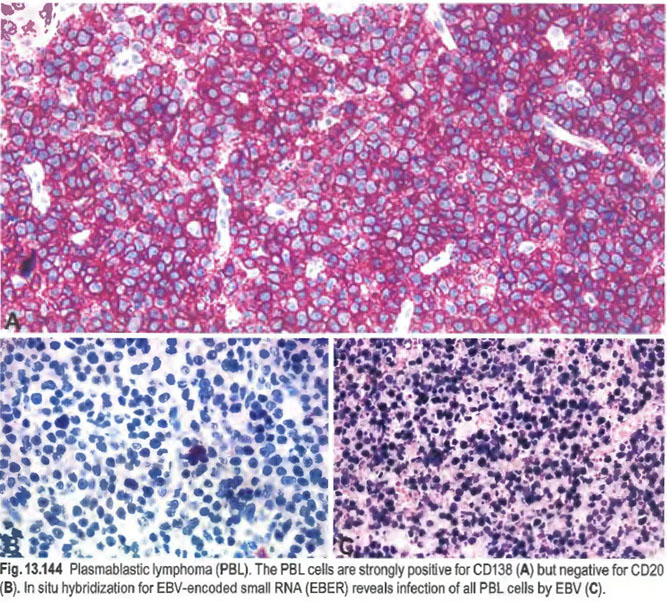
Primary effusion lymphoma (PEL)
B-cell ca that presents c (usually pleural, also pericardial or peritoneal) effusion and not as a mass
- assoc c HHV8 and co-infx c EBV, thus usually in immunocomp pts c HIV esp those c Kaposi sarcoma
- also in older adults (that aren't immunocomp) in endemic areas
- also assoc c multicentric Castleman's
Micro: highly variable, can look plasmacytoid or have RS-like cells
IHC: (+) CD45, ORF73 LNA-1 (LANA)
- negative pan-B markers (CD19/20/79a), T markers (CD10/3/5, and myeloid ags (CD13/14/33), BCL6
- EBER ish usually (+), but EBV LMP1 usually negative (also, EBV not necessary for PEL pathogenesis)
Genes: clonal / hypermutated Ig genes
Px: very poor (< 6 mo)
PEL cytology, prominent nucleoli and a plasmablastic appearance [7]

Extracavitary primary effusion lymphoma (PEL) presenting initially as a mass in the large intestine of an HIV-positive patient. A Plasmablastic/anaplastic cells infiltrating between the glands of a large bowel mass. B The nuclei are strongly positive for HHV8 with an antibody to LANA1 (also called ORF73). [7]

HHV8-associated lymphoproliferative disorders
In addition to causing Kaposi sarcoma, HHV8 (aka Kaposi-sarcoma-associated herpesvirus) can cause a spectrum of lymphoprliferative disorders, including:
- HHV8+ multicentric Castleman disease (MCD)
- HHV8+ DLBCL, NOS (frequently occurs in the background of MCD)
- Germinotropic lymphoproliferative disorder (GLPD)
Except for GLPD, these diseases occur in the setting of HIV infection and in HHV8-endemic areas, but can occur in other immunosuppressed states, such as after transplant
- GLPD usually seen in immunocompetent individuals

[23]
HHV8-positive diffuse large B-cell lymphoma, NOS
- usually arises with HHV8+ multicentric Castleman disease (MCD)
- characterized by a monoclonal proliferation of HHV8-infected lymphoid cells resembling plasmablasts, expressing IgM lambda
- usually associated with HIV infection
Micro: cells resemble plasmablasts and have abundant cytoplasmic immunoglobulin
The neoplastic cells correstpond to a naive, IgM-producing B-cell without IG somatic hypermutations
- must be distinguished from plasmablastic lymphomas in the oral cavity and other extranodal sites that frequently show class-switched and hypermutated IG genes
DDx: Primary Effusion Lymphoma (PEL) -- HHV+ DLBCL NOS is EBV-negative, does not have IG gene mutations, and are thought to arise from naive IgM lambda-positive B-cells (rather than terminally differentiated B-cells)
Leukaemic HHVB-positive diffuse large Bcell lymphoma, NOS, arising in multicentric Castleman disease. Wright-Giemsa-stained tumour cells in the peripheral blood.

HHVB-positive diffuse large B-ce ll lymphoma, NOS, arising in multicentric Castleman disease. A Sheets of plasmablasts efface normal lymph node architecture. B High magnification showing sheets of neoplastic plasmablasts. C Tumour ce lls are negative for immunoglobulin kappa light chain. D Tumour cells are positive for lambda light chain indicating lambda light chain restriction. [7]
HHV8-positive germinotropic lymphoproliferative disorder (GLPD)
Monotypic HHV8+ LPD that occurs in HIV-negative pts
- HHV8+ plasmablasts partially or completely replace germinal centers
-- the plasmablasts show either kappa or lambda light chain restriction but are polyclonal or oligoclonal
- Co-infection with EBV is characteristic
Micro: overall retention of nodal architecture; lymphoid proliferation is characterized by medium-sized to alrge lymphoid cells resembling plasmablasts that involve or replace germinal centers
IHC: large plasmablastic cells are always positive for HHV8 (LANA1), and usually positive for EBER
- rarely can be EBER negative
- plasmablasts negative for CD20, CD79a, CD138, BCL6, and CD10
- CD30 -/+
- negative for EBV latency proteins LMP-1, EBNA2, and BZLF-1 indicating latency 1
Px: usually a favorable response to chemo/radtation

HHV8-positive germinotropic lymphoproliferative disorder. A Germinal centre replaced by confiuent sheets of plasmablasts. B Germinal centre largely replaced by plasma blasts. C Plasmablasts are seen within a hyperplastic germinal centre. D Clusters of plasma blasts within hyperplastic germinal centres. [7]
Burkitt lymphoma
B-cell neoplasm with very short cell multiplying times that usually presents at extranodal sites or as an acute leukemia, with uniform small round blue cells
- numerous mitotic figures and apoptotic bodies --> "starry-sky" pattern
- MYC translocation is very characteristic of this dz, but not specific
- may derive from memory B-cells
Has at least 3 different clinical variants:
Endemic (African) - young males in equitorial Africa, esp in jaw
- EBV virus present in nearly all cancer cells; also strong link to malaria
Sporadic (Western) - may be a large % (up to 1/2) of childhood lymphomas; can occur in adults; jaw tumors rare, is usually abdominal;
- occasionally assoc c EBV (up to 30%)
Immunodeficiency-assoc (Burkitt-like lymphoma) - assoc c AIDS; occurs early in infx in LN when T cells aren't yet depleted
Micro: diffuse prolif of med round nuclei c multiple small nucleoli (up to 5 nucleoli each); tumor cells can mold to one another, has dark blue with lipid vacuoles; lots o mits; tumors can surround LNs,
- "Starry sky" pattern due to tingible body macrophages ingesting tumor cells amongst a sheet of tumor cells
- sometimes (esp in the immunodeficiency types) can see plasmacytoid cells
IHC: (+) CD19/20 (bright)/PAX5/22/10 (strong)/38/77/43, BCL6 (variable, but should have some positivity), Oil Red O (stains lipid vacuoles), sIg (esp IgM) and surface light chains, C-myc
- negative CD5/23/34, BCL2 (in 80%), Tdt, PAS
- very high Ki67 (~100%)
- should only have a few non-neoplastic T-cells
- variable proportion of cases are EBV-driven (EBER+) [11]
- MYC IHC in BL -- clone Y69, epitomics, Burlingame CA; BL typically shows 90-100% tumor cells staining of high intensity; setting a high cutoff (>70% tumor cell staining) may be 100% sensitive and 93% specific for the presence of a MYC rearrangement (although should still do it by FISH)
Genes: t(8;14) MYC translocation / rearrangement at 8q24 to IG heavy region 14q32 (in 90%)
- can also see t(2p12;8) MYC to kappa or t(8;22q11) MYC to lambda Ig
- TCF3 or ID3 mutations seen in up to 70% of cases
DDx: Benign reactive process
-- florid reactive follicular hyperplasia (can look scary, especially in tiny specimens like FNA and small core biopsies, both can have a starry-sky appearance) -- try to see clonality on flow (although single germinal centers can have clonality)
DDx: Lymphoblastic Lymphoma (are TdT+, surface Ig-, and no MYC rearrangement)
DDx: Blastoid variant of mantle cell lymphoma (CyclinD1+, no MYC rearrangement)
Tx: fairly good response to chemo (the chemo regimen is aggressive)
b/c pts usually present at advanced stages, tumor lysis syndrome is common (esp in Burkitt leukemia [MC in males])
Px: poor c BM/ CNS involvement, >10 cm tumors, high LDH
- a prominent histiocytic response usually indicative of better px

Burkitt Lymphoma

Burkitt Lymphoma types [11]

(terms are no longer used)
Classic IHC in Burkitt (clockwise from top left): CD20+, BCL2-, Ki67 100%, CD10+, BCL6+

MYC IHC in BL - assoc c MYC rearrangement
Burkitt lymphoma smear. Notice the dark blue cytoplasm and the cytoplasmic vacuoles
BL - by itself, MYC does not correlate with high Ki67
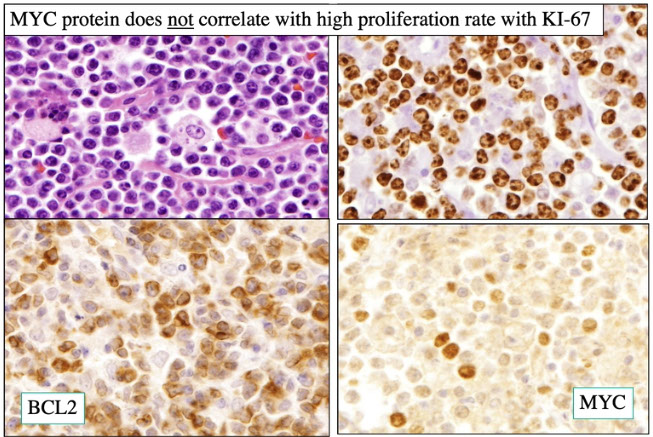
Burkitt-like lymphoma with 11q q aberration
New provisional entity that closely resembles Burkitt lymphoma but lacks MYC rearrangement and has some other distinctive features
- more likely to occur in LN c less MYC expression, more complex karyotypes, cytologic pleopmorphism, and may have a follicular pattern
Genes: cr 11q alteration characterized by proximal gains and telomeric losses
- detection by array CGH
- often have more complex karyotypes than BL
Px: Similar clinical course to BL, based on few cases reported
- some develop post-transplant

(E-H) Burkitt-like lymphoma with 11q aberration. (E) The touch imprint demonstrates a monotonous populatio of transformed cells with basophilic cytoplasm that are (F) CD201, (G) have a very high MIB1/Ki-67 proliferation fraction, and are (H) BCL61. (A-B) Hematoxylin and eosin stain; (C,D-H) immunoperoxidase stains as specified; (E) Romanowsky-type stain.


High grade B-cell lymphoma
High grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements
Cell of origin (COO)
–Germinal center B-cell phenotype (GCB) in most cases
–Minority are non-GCB (MYC + BCL6 rearrangements)
MYC expression by IHC
–High (>40%) in most cases
–Minority have low MYC expression
•Aggressive clinical features.
•Cytogenetics:
–Presence of MYC rearrangement (8q24) and BCL2 (18q21) and/or BCL6 (3q27) rearrangement
–“double-/triple-hit lymphoma”
•Morphology:
–DLBCL, NOS
–BCLU (closer to BL than DLBCL)
–Blastoid appearance
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma - DOES NOT EXIST ANYMORE
- this entity has been replaced with High grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements and High grade B-cell lymphoma, NOS
IHC: (+) CD45, B-cell markers, CD10, Bcl6, high Ki67,
- neg: Bcl2, MUM1
HGBCL with MYC and BCL2 and/or BCL6 rearrangements morphological variation


Survival in HG BCL with MYC and BCL2 and/or BCL6 rearrangements
High-grade B-cell lymphoma, NOS
Aggressive mature B-cell lymphoma
•Morphology:
–Burkitt lymphoma-like
(BCLU in 2008 WHO)
–Blastoid cytology
•Cytogenetics:
–Absence of MYC rearrangement and BCL2 and/or BCL6 rearrangement
•Heterogeneous category
High grade B-cell lymphoma, NOS

High-grade B-cell lymphoma, NOS- has blastoid morphology, TdT-, no double/triple-hit genetics (although MYC is rearranged)

B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical Hodgkin lymphoma
aka Mediastinal gray zone lymphoma (GZL)
- is a mix bwt nodular sclerosis classical Hodgkin lymphoma and DLBCL (esoecially primary mediastinal [thymic] large B-cell lymphoma) [1]; suggesting biologic coneection bwt the two
- usually in young males (20-40 yo)
May have morphology that looks like cHL-NS but with IHC suggesting PMBL
____________________________________
IHC: (+) CD45, B-cell markers, Bcl6 (var), CD30/15
- neg: CD10
____________________________________
DDx: PMBL
Common features of Primary Mediastinal (Thymic) Large B-cell Lymphoma (PMBL) and CHL-NS [10]
- F>M; 2:1 ratio
- adolescents, young adults
- Mediastinal mass +/- supraclavicular LN often involved
- Common cytogenetic alterations -- gains at 9p24 (JAK2) and 2p16 (REL)
- Similar gene expression profile with activation of NFKB pathway
- thought to arise from a "thymic B-cell"
- Borderlands between Hodgkin's lymphoma and PMBL still exist
- Mediastinal Gray Zone Lymphoma (GZL) may represent the "missing link" between CHL-NS and PMBL
- gray zone lymphomas exhibit a morphological and immunophenotypic continuum
- MGZL cannot be readily classified as either PMBL or CHL, with frequent asynchronous histology and immunophenotype
- Clinical features similar to NS-CHL and PMBL, except for male predominance
- Composite and sequential lymphomas are a related phenomenon
- the distinction between CHL-NS and DLBCL (PMBL) is still important as there are some major treatment differences [10]
____________________________________
Tx: NCI protocol initiated, using regimen sucessful in PMBL, DA-EPOCH R, with mediastinal radiation for those PET+ at conclusion of Rx
- PD-1 blockade is a new therapeutic tool in the treatment of CHL, PMBL, and GZL
- amplification at 9p24.1 leads to increased expression of PD-1 ligands, PD-L1 and PD-L2 on the neoplastic cells
- associated with enhanced KAJ-STAT signaling and expression of pSTAT3
- PD-1 blockage with nivolumab or pembrolizumab is effective in CHL patients who have failed standard therapy [Ansell et al. NEJM 2015. Armand et all JCO 016]
____________________________________
Px: worse than both DLBCL (PMBCL) and cHL
GZL. The thymic B-cell, there are influences that direct the fate of the thymic B-cell that are probably a combination of epigenetic, genetic, and environmental factors

Gray zone lymphoma (GZL) between CHL-NS and PMB. Upper left shows sheets of neoplastic cells that are negative for CD20 but positive for CD15 and CD30. Qualifies as a GZL bc morphologically looks like a DLBCL but by IHC stains as CHL

Another case of MGZL, which looks like CHL by morphology, with background inflammatory cells, but shows sheeting out of HRS-like cells, the B-cell program is retained (CD20+) , CD15-, OCT2+, this is the most common pattern in GZL
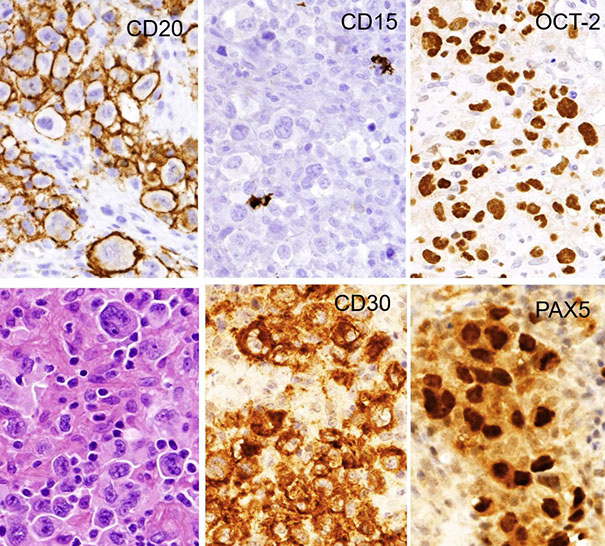
RNAseq PMBL, NSCHL, and MGZL, showing that the cases of GZL were overall closer to CHL than to PMBL

Mature T and NK-cell Neoplasms
1/20 lymphoid neoplasms, highest in Asia; Welsh and Irish get enteropathy assoc T cell lymphoma
- PTCL NOS > AITCL > ALCL > ATCL

Survival curves of T-cell neoplasms [Vose et al. JCO 2008]
WHO Classification of T/NK cell neoplasms [12]

T-cell prolymphocytic leukemia (T-PLL)
A rare, aggressive T-cell cancer with post-thymic T-cell IHC affecting PB, BM, LNs, HS and skin in adults (avg 65 yo)
- labs: anemia, TBCpenia and marked leukocytosis; HTLV-1 negative
Micro: commonly has cytoplasmic blebbing / "pseudopod-ish"
IHC: (+) CD2,5,7,3 (weak), 52 strong;
-- 60% are CD4+ and CD8-; while 25% are CD4/CD8 double+, which is almost exclusive for T-PLL; 15% are CD4-/CD8+
- negative Tdt, CD1a
- T-cells stain strongly c a-naphthyl acetate esterase and acid phosphatase c dot-like staining in Golgi area
Genes: TCR clonally rearranged
Inversion 14 c q11 (in 80%) and q32 breakpoints
- 1/10 c reciprocal tandem t(14;14)(q11;q32)
- activate the oncogenes TCL1A or TCL1B by moving it next to the TRA@
May be related to Ataxia Telangiectasia, as the Ataxia Telangiectasia Mutated (ATM) gene shows missense mutations
Px: aggressive; survival 1-2 yrs

T-PLL
T-cell large granulocytic lymphocytic leukemia (T-LGL)
Large granular lymphocytes (LGL) constantly increase in PB over period of >6 mo; can be Tc cells or NK cells
- M=F, older adults
Labs: Neutropenia, anemia, but no TBCpenia
Criteria:
- LGLs > 2 x 10^9/L in blood
– > 6 months
– No other cause (infxn, drug)
Tc LGL leukemia assoc c neutropenia Smegaly, polyclonal hypergammaglobulinemia; may arise in setting of prolonged immune stimulation (is assoc c autoimmune dz [RA])
- absent homeogenic apoptosis
- may be diagnostically difficult after BM transplant
- have TCR rearrangement
- usually indolent; more aggressive if CD56+ c blastoid morphology
NK cell LGL leukemia has neutropenia, anemia, fever, jaundice, and HSmegaly
- is EBV negative (vs aggressive NK cell leukmeia [ANKL]
Micro: the lymphocytes look large with lots of fairly transparent cytoplasm which contains several granules in "parallel tubular arrays"
- BM usually slightly hypocellular, left-shifted, and fibrosed, hard to see the LGLs
IHC: (+) CD3/8, TCR-ab (95%, 5% TCR dg), aberrant NK markers CD57 and CD16 (80%), CD94/NKG2/KIR (50%+), cytotoxic markers (TIA1 granzymes B and M),
- diminished or lost CD5/7
- granules are acid phosphatase and B-glucuronidase positive (though people dont really use these stains)
Genes: Must have TRG gene rearrangement
- 1/3 of cases with STAT3 mutations
Px: indolent, non-progressive
- morbidity caused by cytopenias (esp neutropenia)
- patients with significant morbidity may be treated with immunosuppressive agents
- STAT3 mutations does not effect outcomes
Chronic lymphoproliferative disorder of NK cells (CLPD-NK)
- Provisional entity
- Diagnosis requires large granular lymphocytes (LGLss) > 2 x 10^9 for greater than 6 months with no other reactive or neoplastic cause determined
- cytopenias common in patients CLPD-NK or T-LGL, usually neutropenia
IHC: aberrant expression patters in NK-cells can be detected, however, may need to analyze KIR receptors such as CD157a, b, and e
Genes: STAT3 mutations in 1/3 (also seen in T-LGL, suggesting a common pathway leading to disease)
Tx: similar to T-LGL, many patients do not require treatment for many years
Systemic EBV+ T-cell disorders [23]

Aggressive NK cell leukemia (ANKL)
NK-cell cancer almost always assoc c EBV
- Rare; younger (42 yo) Asians, M=F
- MC in PB, BM, liver, spleen, presenting c fever and leukemic blood sx, c decrease in main lineages
- labs: elevated LDH
Micro: highly variable
IHC: (+) CD2/c3/56/16(in 75%), cytotoxic molecules, FAS ligand, EBV
- negative CDs3/4/5/34/57, Tdt
Px: most have fulminant course c multiorgan failure
- <2 mo survival
Ebstein-Barr virus (EBV) positive T-cell lymphoproliferative diseases of childhood
-
Systemic EBV+ T-cell lymphoma of childhood
- usually primary - may progress from CAEBV, systemic or cutaneous
- more common in children, but can occur in adults
- acute presentation, often following primary EBV infection
- variable cytological atypia, but more than seen with CAEBV
- HLH-like picture is common

Systemic EBV+ T-cell lymphoma of childhood. Diffuse infiltrate of the paracortex with focal residual lymphoid follicles surrounded by EBER+ lymphoid cells
Systemic EBV+ lymphoma of childhood. See that the cells show considerable cytologic atypia , CD3+ and perforin+. Showed a T-cell gene rearrangement

Chronic active EBV (CAEBV) infection of T- and NK-cell type, systemic form
A systemic lymphoproliferative disorder derived from T-cells or NK-cells
- can be polyclonal, oligoclonal, or monoclonal
Clinical symptoms include fever, hepatitis, pneumonitis, cytopenias
- hepatosplenomegaly and lymphadenopathy
Persistence of symptoms for >3 months, following acute EBV infection
- Evidence of EBV in affected organs, and increased EBV viral load
Liver biopsy from a patient with CAEBV showing focal hepatocellular necrosis and bland-appearing lymphoid infiltrate, mainly sinusoidal in distribution

CAEBV, immunohistochemistry, showing lymphocytes are cytotoxic T-cells, only infrequent B-cells seen iwth CD20, mostly in portal aggregates

CAEBV, EBV stains over time, TCR gene rearrangement negative, indicating indolent clinical course

Hydroa vacciniform-like lymphoproliferative disorder (HV-LPD)
- the term "HV-LPD" reflects the range in pathological features without sharp distinctions between HV and HV-like lymphoma
- most often in Asian or Hispanic children
- lesions in sun-exposed areas
- papulo-vesicular eruption that proceeds to ulceration and scarring
- some patients may have evidence of systemic CAEBV
IHC; variable with cells being of either T-cell or NK-cell origin
- most cases are ab (CD8+) but cases with a gd T-cell phenotype are seen
Genes: Clonal TCR rearrangement common in T-cell cases
- NK-cell origin in up to one-third
Px: chronic course, often remits in adult life
- may proggress to acute phase with systemic disease
- systemic disease more common in Asian / Hispanic populations than in white
[Quintanilla-Martinez, L Blood 2013]
HV-LPD. Typical case, showing an infiltrate in the dermis, note there are many more infiltrating T-cells than there are EBV cells

HV-LPD with a more vesicular type of eruption. Mild perivascular lymphoid infiltrate with relatively small number of EBER+ cells

HV-LPD. Some cases show clinical and histologic progression over time. This patient had skin lesions for over 21 years, that graually progressed. Here see a dense dermal infiltrate and sheets of EBER+ cells, which was associated with clinical progression

Severe mosquito bite allergy
- EBV+ NK-cell lymphoproliferative disorder with high fever and intense local skin symptoms, including erythema, bullae, ulcers, skin necrosis, and deep scarring following mosquito bites
- pts have NK cell lymphocytosis in PB and inc risk of HLH and progressing to overt NK/T-cell lymphoma or aggressive NK cell leukemia
- very uncommon, usually in Japan, China, or Mexico
Adult T-cell leukemia / lymphoma (ATCL / ATLL)
Peripheral T-cell cancer with pleomorphic lymphoid cells caused by Human T-cell Leukemia Virus type I (HTLV-1)
- endemic in Japan, Caribbean and central Africa, where HTLV-1 is high; may be spread through breast mild, has long latency period (avg age 58) where it mutates the HTLV-1 basic leucine zipper factor (HBZ)
Clinically, presents with widespread LN and PB involvement (usually spares the BM), usually c skin rash (>60%), hypercalcemia, lytic bone lesions (30%), lymphadenopathy, hepatosplenomegaly, nearly 100% are stage IV
- patients are susceptible to opportunistic infections in the absence of therapy or granulocytopnenia (Pneumocystis carinii pneumonia, Cryptococcal meningitis, cytomegalovirus infections, candida, strongyloides, mycobacterial infections)
Treg cells are another CD4+ T-cell subset which shut off and suppress immune responses; thought to prevent autoimmunity
- have high density of CD25 and transcription factor FOXP3, and also CD4
- T cell leukemia/lymphoma (ATLL) has been linked to Treg cells through CD25 and FOXP3 expression, and also explains the immunospreesion assoc c this dz
- The hypercalcemia in ATLL has been linked to osteoclast-activating activity
Variants
1. Acute--leukemic (esp in pts from Japan)
2. Lymphomatous variant— lymphadenopathy
3. Chronic variant— Lymphocytosis (WBC increased), ATL cells >10%, mild LAD, slight HSmegaly, skin lesions variable, LDH slightly inc, Ca++ normal, survival usually >2 years
4. Smoldering variant-- WBC normal, ATL cells <3%, no LAD, no HSmegaly, skin rash with erythema and papules, Normal LDH and Ca++, survival >2 years
__________________________________________
Lab: hyperCa2+, with/without lytic bone lesions
__________________________________________
Micro: acute variant has poly-lobated cloverleaf cells or "flower cells"; whereas the the chronic type has smaller "flower-oid" cells with nuclear indentations and convolutions
Architectural effacement, paracortical distribution, lymphoma cells mostly small to intermediate in size with mature chromatin and irregular nuclear contours
- Scattered and loosely clustered large cells, Hodgkin-like
- skin lesions (found in >1/2 pts) have Pautrier-like microabscesses
- RS-like cells may be present
__________________________________________
IHC: (+) CD2 / 3 / 5 / 4 / 25, CD30 (variable, in larger cells), CCR4, FOXP3+/-, BF1, TcR rearrangements
- 10-15% are CD4/CD8+, can be CD4-/CD8-
- cells are phenotypically and functionally Treg cells, which function as suppressors of immune function (correlates with immunodeficiency characteristic of ATLL)
Small to medium sized cells:
+ CD3, CD4, variable CD25
- CD8
Hodgkin (HRS)-like cells:
+ CD20, PAX5, CD30 (heterogeneous), EBER
- CD15
- negative CD7 / 8 / 56 (var), cytotoxic markers (TIA1, granzyme, perforin)
__________________________________________
Genes: clonally integrated HTLV-1, rearranged TCR genes
__________________________________________
Tx:
Lymhomatous - 7 drug regimen, toxic, 100% neutropenia
Acute - antiviral first line at UM, AZT, IFN-a
Targeted - anti- CD25, CCR4, CD30, CD52
__________________________________________
Px: acute and lymphomatous forms have poor px, smouldering the best, chronic less so
- age, serum Ca, LDH and performance status all important in px
- death usually comes by opportunistic infx
ATLL. Polylobated "flower" cells

ATLL. Diffuse involvement of LN, polylobated cells in the lymphatic sinuses.

ATLL. can have RS-like EBV+ cells, similar to AITL and Hodgkin lymphoma, which can be PAX5+ and negative for T-cell markers

ATLL types

Cutaneous involvement of ATLL, can look like Pautrier microabscess of MF.

ATLL skin lesions. May be ulcerated.

ATLL. Polymorphous cells, sometimes multilobulated cells.

ATLL. May look like DLBCL
Smoldering ATLL bx of skin rash, non-specifc perivascular inflammation

Smoldering ATLL skin rash, appears hyperkeratotic
Smoldering ATLL, CD3+, CD25+

NTNKT
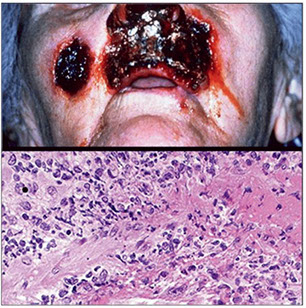

Extranodal NK-T-cell lymphoma, nasal type (NTNKT)
- aka polymorphic reticulosis, lethal midline granuloma, angiocentric lymphoma
EBV-assoc extranodal lymphoma c lots of necrosis, vascular damage
- can occur anywhere in upper respiratory tract (usually in the nose) and also (possibly from dissemination) in GI, testis, skin, STs
- associated with increased risk of hemophagocytic syndrome`
Micro: dense lymphocytic infiltrate that destroys glands and vessels (angioinvasive/ angiodestructive) and causes lots of necrosis
- intracytoplasmic azurophilic granules seen on touch preps
IHC: (+) CD3 (cytoplasmic) / 56 / 2, EBV, Latent Membrane Protein (LMP), cytotoxic markers (perforin, granzyme, TIA1)
- neg: surface CD3, CD5 / 4 / 8 / 30 (var), BF1, TcR rearrangements
Px: some respond well to tx, some expire despite aggressive tx
- fairly poor outside nasal cavity vs those confined to the nasal cavity


Lesion of the nasal mucosa with polymorphous infiltrate that could be mistaken as a reactive process

Enteropathy-associated T-cell lymphoma (EATL)
HG TCL in pts c longstanding celiac disease, usually preceded by refractory sprue c mucosal ulceration (ulcerative jejunoileitis); Welsh and Irish MC affected
- assoc c HLADQA1*0501 and DQB1*0201 genotypes
Often presents with intestinal perforation
__________________________________________
2 flavors:
Type 1 (4/5) : "classic variant"
- not designated as Enteropathy Associated TCL (EATL)
Rare, pleomorphic lympoma assoc c celiac sprue, dermatitis herpetiformis, hyposplenism, seen in Northern Europeans; 95% of patients with HLA-DQ2 and HLA-DQ8
- autoantibodies against tissue transglutaminase, and antibodies against gliadin (gluten-free diets may reduce the risk of lymphoma)
- IHC: (+) CD3, 30, CD103, TIA-1, usually ab
- negative CD4,5,56, CD8 (+ in 1/5)
Type 2 (1/5)
- now called monomorphic epitheliotrophic intestinal TCL (MEITL)
Small/medium monomorphic cells with clear cytoplasm, usually gd
- no assoc c celiac dz, inc incidence in Asians and Hispanics
- IHC:(+) CD8/56, TIA1, MATK, often gamma-delta+ (though not necessarily)
- aberrant CD20 in 20%!!
-- CD4-, CD5-
__________________________________________
Micro: Broad morphologic spectrum
- the adjacent mucosa shows villous atrophy
__________________________________________
Genes: STAT5B in 36% of type II
__________________________________________
Px: agressive clinical course with poor prognosis

(E) EATL. The somewhat pleomorphic intestinal infiltrate extends into the epithelium and would be associated with enteropathic changes elsewhere in the intestine. (F) MEITL.The monotonous intestinal infiltrate is very epitheliotropic (CD8 immunoperoxidase stain )
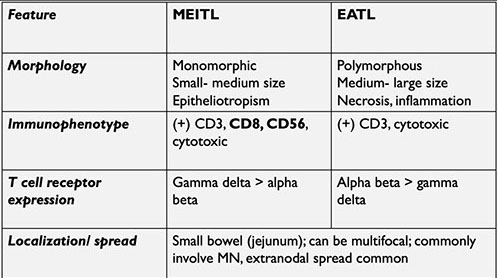

EATL

EATL. Villous atrophy and flattening, with increased lymphs

HSTL
Hepatosplenic T-cell lymphoma (HSTL / HSTCL)
- aka late-onset post-transplantation lymphoproliferative disorder of host origin (catchy)
systemic cancer derived from functionally immature cytotoxic gd (gamma-delta) Tc cells with Vd1 usage that infiltrates sinusoids of Hepatos and Splenos and marrow
- rare (<1% nHL), M>F, mostly in immnocompromised (those with Crohn's disease on anti-TNF tx, late-occuring PTLD) in young males c B sx;
- CD8+ Tc cells have gd TCR and isochrome 7q
- do NOT have LAD, and is NOT assoc c EBV
- ab (alpha-beta) type in females and has wider age distribution
- Bone marrow usually involved at time of dx
__________________________________________
Micro: red pulp diffusely involved c monotonoous cells c small rim of light cytoplasm
- in BM see tumor cells in sinusoidal distribution
__________________________________________
IHC: (+) CD2/3, TCR-delta, CD56,
- has nonactivated cytotoxic profile: TIA1+, Granzyme B negative, perforin negative
- negative TCRab, CD4, CD8, CD5, granzyme B, Tdt
Rare cases ab, similar clinicopathologic and molecular features
__________________________________________
DDx: T-LGL in BM, spleen
__________________________________________
Genes: TRG@ rearrangement, isochromosome 7q
- STAT5B (mutated in 35%) and STAT3 in HSTCL and primary cutaneous TCL, and gamma delta T-cell lymphomas of the GI tract (?[7])
- SETD2 in 25%
May have +8 and -Y
__________________________________________
Px: Aggressive, <2 yr survival
HSTL in spleen. White pulp is atrophic / absent. Red pulp expanded with atypical cells filling the siniusoids

HSTL, may also see hemophagocytosis

HSTL characteristic IHC. CD5-, CD56+, TCRg+, TIA1+

HSTL in the liver IHC.

Subcutaneous panniculitis-like T-cell lymphoma (SPTCL)
Rare Tc cell neoplasm with multiple subQ nodules
- pleomorphic lymphs, karyorrhexis of tumor cells and fat necrosis in subQ; HSmegaly can be there but LNs are not usually involved
- can look clinically like lobular panniculitis
- broad age range (1 - 57 years), median 30 yo
Males = Females
- Deep subcutaneous nodules, primarily affecting extremities, trunk [12]
__________________________________________
-Labs: cytopenias, inc LFTs, hemophagocytic syndrome
- is gd T-cell receptor negative (if positive, call it primary cutaneous gd T-cell lymphoma)
- is EBV-negative
__________________________________________
Micro: inflam in fat lobules, sparing the septa and also not the dermis or epidermis (usually confined to the subcutis)
- see rimming of cancerous T cells around fat cells (characteristic but not specific)
- necrosis and karyorrhexis prominent
- primarily lobular distribution, with or without septal infiltration
__________________________________________
IHC: (+) [ab T-cell phenotype] CD3/8, cytotoxic moldecules (granzyme B, perforin, TIA1), BF1, TcR gene rearrangements
Have an activated ab T-cytotoxic phenotype [12]
--- CD3+, CD8+;
--- TIA1+, Granzyme B+, Perforin+,
--- CD56 negative (in contrast to gamma-delta)
-- Negative EBV
--- TCR g/b genes rearranged
- negative CD4 / 56, EBV
__________________________________________
Genes: Germline mutations in HAVCR2 encoding TIM-3
- mutations more common in Asian than European patients
- associated with HLH positive cases
__________________________________________
DDx: Lupus profundus --- mixture of T-cells, B-cells, plasma cells, lobular pattern with preserved septa, fibrinoid change in connective tissue, interstitial infiltration, but infrequent rimming of fat spaces, mixture of CD4+/CD8+ cells, scattered gamma-delta T-cells, and increased plasmacytoid dendritic cells (PDCs) (CD123+) [12]
- not always an easy distinction: 19% of pts with SPTCL ab had history of autoimmune disorder (SLE, RA, ITP), oligoclonal T-cell populations can be seen in some patients with lupus, inclusive of the cutaneous lesions; must correlate clinical histological and genetic features
- one should be cautious about making the diagnosis of SPTCL in a patient with lupus
__________________________________________
Tx: steroids
__________________________________________
Px: >80% at 5 yrs, worse c hemophagocytic syndrome
- rarely goes to LN
SPTCL. A) nodular skin lesion next to area of lipoatrophy; B) subQ infiltrate c rimming of CD8+ T cells around fat

SPTCL sparing the dermis and epidermis. Marked cytologic atypia.

SPTCL - necrosis is common, can see fat necrosis

SPTCL, neoplastic cells rimming the fat. CD8+, TIA1+

Mycosis fungoides (MF)
MCC primary cutaneous T-cell lymphoma (50%), usually in older adults , M>F; is a cutaneous T cell lymphoma (CTCL) that involves LN
- patches to plaques to tumors which is limited to skin
- may have extracutaneous spread, but rarely involves BM
Micro: atypical haloed lymphs c possible cerebriform nuclei infiltrate basal layer of dermis (epidermotropism)
- intraepidermal Pautrier microabscesses seen in plaques
- can histologically transform to large cells
- MF does not have much spongiosis or dying keratinocytes
- collagen is a little bit thicker in MF,
Patch, Plaque and Tumor stage
Patch stage
• Erythematous patches, irregular in size and shape
random distribution, usually sun protected sites
• Mild superficial perivascular lymphocytic infiltrate c
epidermotropism (lymphocytes in the epidermis
without associated spongiosis)
• Lymphocytes lining up at the DEJ (“birds on a wire”)
• Immunos required – Loss of CD7 and CD5 typically
• T-cell gene rearrangement
• *Must have proper clinical presentation
• DDx – chronic dermatidities
Plaque stage
More epidermotropism and Pautrier microabscesses
x
Tumor stage
Gross: previous patch/plaque lesions or biopsy proven MF
– Exophytic, irregular nodules w ulceration
Micro: Often loses epidermotropism;
- Dermal infiltrate c Variable sized T-cells (small, med, large)
Large cell transformation can occur in up to 50% of patients with advanced disease, and is defined by >25% large cells
- CD30+ in large cell-transformed MF has a significantly better outcome than CD30- large cell transformed MF [26]
IHC: (+) CD3/4/45, TCR-B, cytotoxic granules (only in realy patch/plaque), can be CD8+ (TC cell variant)
- negative CD2/5/8/7/20 (loss of T cell ags, loss of CD7 is MC)
- matching T-cell clones from different bx sites can be helpful to dx
DDx: Langerhans cells can also aggregate in the epidermis (but will not be positive for CD3); be careful because Langerhans cells can also be CD4+
Genes: TCR clonal rearrangement
Staging by ISCL-EORTC system (4 stages)
Px: Indolent with slow progression if limited dz, poor px with late dz
MF - epidermotropism
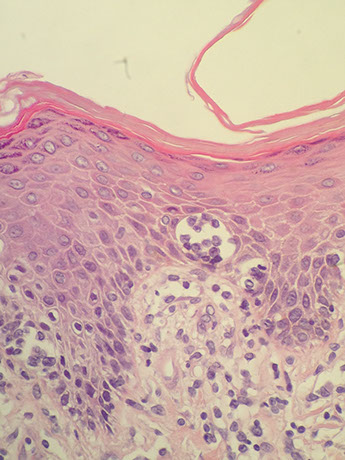
MF - Pautrier microabscess
MF - kinda looks like vacuolar interface dermatitis (MF does not have much spongiosis or dying keratinocytes), collagen is a little bit thicker in MF,

Tumor stage (clinical term) MF, lacks epidermotropism, difficult to know this is MF without history

Folliculotropic MF

MF IHC panel

Sezary syndrome
Rare, <5% cutaneous T-cell lymphomas, though is technically a leukemia
- older pts, M>F, presenting with itchiness, spots of hair loss, ectropion, and the criteria below
Triad:
1. Erythroderma
2. Generalized lymphadenopathy
3. Sezary cells in skin, LN and PB
-also needs one of the following:
a. absolute Sezary cell count of 1000/mm3
b. exoanded CD4+ T-cell population c CD4/CD8 ratio >10
c. loss of one of more T-cell antigens
Micro: infiltrates similar to MF, but can be more monotonous and have less epidermotropism
- often non-specific, sparse BM infiltrates
IHC: (+) CD2 / 3 / 4 / 5
- negative CD8 / 7 / 26
Genes: clonal rearrangements of TCR
- JUNB amplification
Px: poor, 10% @ 5 yrs
- death from opportunistic infx


Sezary syndrome
Primary cutaneous CD30 positive T-cell lymphoproliferative disorders
-
Primary cutaneous anaplastic large cell lymphoma (pcALCL)
Comprised of large anaplastic, pleomorphic, or immunoblastic cells, the majority of which (>75%) express CD30; usually ALK-
Second MCC of cutaneous T-cell lymphoma, median age of 60 years, M:F is 2-3:1
- 80% have solitary lesions
- pcALCL should be excluded if the patient has a hx of MF (in which case, should dx MF with large cell transformation) or if the patient has evidence of systemic ALCL
If no history is given, may render the diagnosis of "CD30-positive T-cell lymphoproliferative disease" with a differential of type C LyP, pcALCL, and secondary skin involvement of systemic ALCL [15]
- these 3 lesions look identical, need to do clinical staging
__________________________________________
Micro: sheets of CD30+ tumor cells, may have epidermotropism (esp in cases with DUSP22-IRF4 rearrangment)
- ulcerating lesions may show a LyP-like histology with relatively few CD30+ cells
__________________________________________
IHC: shows activated CD4+ T-cell phenotype with variable loss of CD3, CD5, and/or CD3 with frequent expression of cytotoxic proteins
- May be CD4-/CD8- or CD4+/CD8+
- Need CD30+ in majority (>75%) of lesional cells by definition
- CD15+ in 40% of cases
- IRF4/MUM1+ in almost all cases
- Negative for PAX5 and EBV
__________________________________________
Genes: most cases show TCR gene rearrangements
- vast majority do not have ALK gene translocations at cr2
- DUSP22-IRF4 rearrangements on cr 6p25.3 seen in 25% of pcALCLs and a small subset of LyPs
- TP63 miutations seen in ~5% of pcALCL
__________________________________________
DDx: LyP; large cell transformation of MF; cutaneous involvement of systemic ALCL; LyP
__________________________________________
Px: favorable, 10-year survival of 90%
- extracutaneous dissemination in 10%, usually in regional LNs
- LN mets and multifocal lesions does not change px
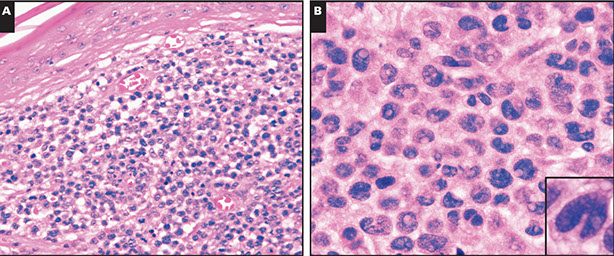
Primary cutaneous anaplastic large cell lymphoma. A-B, Sections show a dermal-based atypical infiltrate that spares the overlying epidermis, composed of large pleomorphic and anaplastic cells with many reniform nuclei [15]
Primary cutaneous peripheral T-cell lymphomas, rare subtypes
Primary Cutaneous gamma-delta T-cell lymphoma (PCGD-TCL)
Rare (1% of T-cell lymphoma), usually in adults, M=F
- may occur in mucosa (where gd T-cells usually live), but usually seen in extremities and has highly variable presentation (can have cutaneous ulcers or plaues, or others)
__________________________________________
Histo: variable, but with epidermotropism
__________________________________________
IHC: (+) CD2 / 3 / 56 / 7 (var), cytotoxic markers (strong +, granzyme, perforin, TIA1), gamma-chain TcR rearrangements
Have an activated cytotoxic immunophenotype
- neg: CD4 / 5 / 8 (rare+), BF1, EBV
__________________________________________
Genes: epidermotropic and dermal are mainly Vd1, and subcutaneous are mainly Vd2
- All show activating mutations in the JAK-STAT pathway [12]
__________________________________________
DDx: subcutaneous panniculitis-like TCL (has involvement of overlying dermis / epidermis
- mycosis fungoides
__________________________________________
Px: poor (resistant to chemo), esp if in subQ fat
- epidermotropic cases resembling mycosis fungoides appear to have a better prognosis
Clinical specturm of Cutaneous gd T-cell lymphomas [12]
PCDG-TCL. Infiltrate is primarily dermal without involvement of the epitdermis

PCGD TCL. Can look a lot like subcutaneous panniculitis-like TCL (SPTCL), however, there is usually involvement of the overlying dermis and epidermis in PCGD.
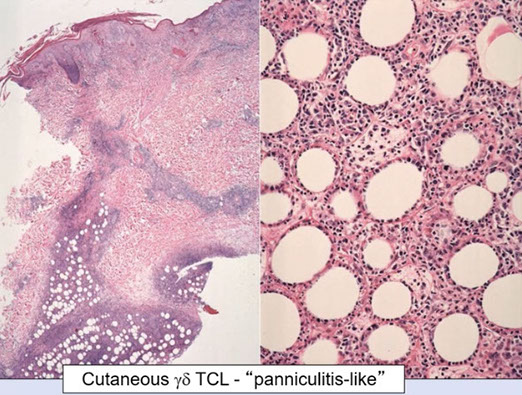
Some cases of PCGD TCL can involve extranodal sites, especially mucosal sites, seen here with gastric involvement

Primary cutaneous CD8-positive aggressive epidermotrophic cytotoxic T-cell lymphoma
Rare (<1% of cutaneous T-cell lymphomas), generalized skin lesions, can disseminate to other organs
- a provisional entity
__________________________________________
Micro: variable
__________________________________________
IHC: (+) CD3 / 8 / 2 (var) / 7 (var) / 45 RA (var), cytotoxic markers (TIA1, granzyme, perforin), BF1, TcR rearrangements
- neg: CD4 / 5 / 30 / 56 / 45 RO, EBV
__________________________________________
Px: poor (2-3 yrs)
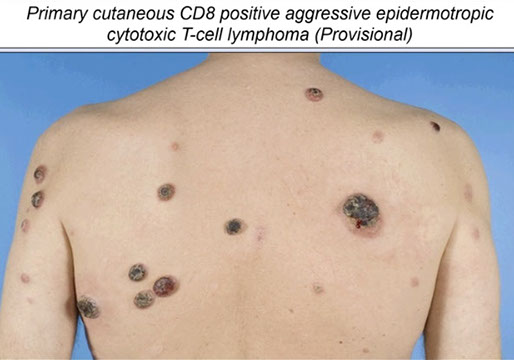
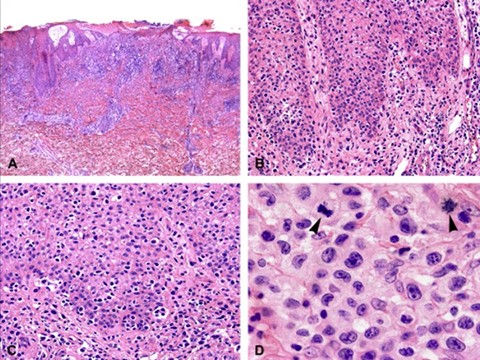



PC CD8+ECTCL (?) - show aggressive histologic and clinical features
PCCD8+ECTCL micro showing epidermotropism with pagetoid-like pattern
Rapid onset generalized plaques and tumors
Low power, shows dense dermal infiltrate with somewhat atrophic epidermis
Dermal infiltrates by lymphocytes with halo, does not really have pautrier microabscess. Dermis shows abnormal cells with dermal edema and extravasated erythrocytes
CD2 is lost. CD4 was also negative
CD3 is strongly positive, extends into those intraepidermal groups. CD8 was also positive
CD7 is positive, which would be atypical for the most common cutaneous T-cell neoplasm (MF)
Perforin is weak, but is diffusely positive
1 - 9
<
>
Indolent T-cell lymphoproliferative disorder of the GI tract
Made of CD8+ T cells
Indolent clinical course
Primary cutaneous acral CD8+ TCL
Made of CD8+ T cells
Indolent clinical course

(C-D) Primary cutaneous acral CD81 TCL. (C) Nodule on the ear. (D) There is a diffuse monotonous infiltrate of CD81 T cells.
Primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder (PC-SMTLPD)
Rare (~2% of cutaneous T-cell lymphomas) that does not have the lesions that one may see in MF, but usually single red papule on the head/neck or upper trunk
__________________________________________
Micro: dense diffuse nodular infiltrates
- can have focal epidermotropism, but not as much as MF
__________________________________________
IHC: (+) CD3 / 4, CD279/PD1, BF1, variable BCL6, TcR gene rearrangements
- can have loss of pan T-cell markers
- may have mix of CD8+, B-cells, and plasma cells which can make diagnosis difficult
- neg: CD8 / 56 (rare+), cytotoxic markers (TIA1, granzyme, perforin), CD30, EBV, low Ki67 (if Ki67>20%, you should question the diagnosis)
__________________________________________
DDx: distinction with atypical hyperplasia often difficult, as lesions sometimes contain numerous B-cells, think PCMZL [19]
__________________________________________
Px: not too bad (esp if single lesion), 4/5 alive at 5-years
- just need local therapy and not systemic
- may need more aggressive tx if bulky disease

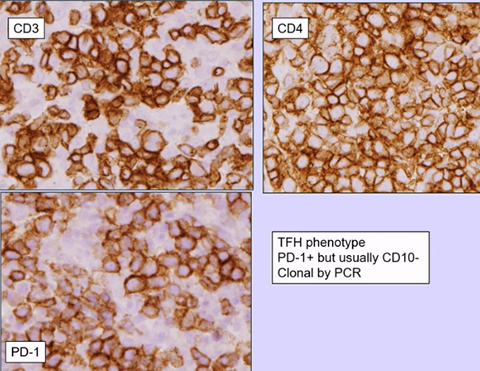
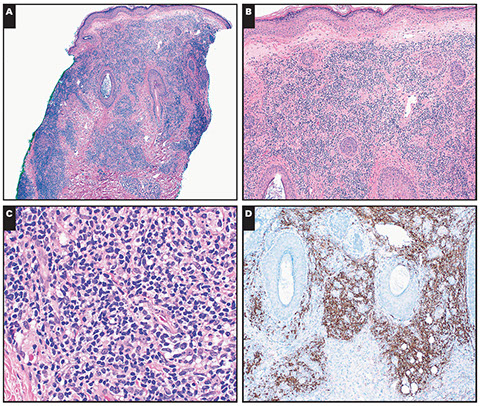

PC CD4+ SM TCLPD (not lymphoma) presenting as a single head lesion
PC CD4+ smTCL
CD4+ TLPD immunohistochemistry
Primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder presenting as a solitary erythematous patch on the right temple of a 52-year-old man. A, A dense, diffuse, and nodular infiltrate extends from the superficial to deep dermis. B, A grenz zone is present. C, The infiltrate is polymorphous, composed of small- to medium-sized lymphoid cells with somewhat irregular nuclear contours, small round lymphocytes, histiocytes, and few plasma cells. Many admixed CD20-positive small B cells (D) are present
1 - 4
<
>
Peripheral T-cell lymphoma, NOS (PTCL)
A diagnosis of exclusion, by definition a heterogeneous category [12]
- characterized by a broad morphologic spectrum
- new approaches include segregation of tumors of TF origin (follicular variant)
- gene expression profiling recognized tumor of TH1 and TH2 origin
- aka the "diffuse large B-cell lymphoma" of the PTCLs (end point of transformation of cells from diverse origins) T-cell neoplasms end up here)
Includes nodal and extranodal T-cell lymphomas that don't fit in other categories, can occur anywhere and infiltrate hematopoietic orgabs
- MC peripheral T-cell lymphomas (1/3 of total)
- rare in kiddos, 2M>1F, BE VERY WEARY OF MAKING THIS DIAGNOSIS IN KIDS / YOUNG ADULTS
__________________________________________
Peripheral T-cell lymphoma, NOS, EBV+
- an uncommon form of PTCL in which tumor cells are EBV+
- monomorphic cytology, lacks necrosis and angiodestruction of EBV+ T/NK nasal-type
- more common in setting of decreased immunity [Attygalle et al. EAHP/SH, 2014]
- Elderly, HIV
- still classified as part of PTCL, NOS
__________________________________________
Hist: very variable, but can see diffuse prolif of small and large lymphs c cloverleaf nuclei, admixed eos, plasma cells, histios, and can have prominent postcapillary venules
__________________________________________
IHC: (+) CD3 / 4 / 56 (var), BF1, TcR rearrangements,
- GATA3 positivity may confer a worse px
- neg: CD8, CD25, cytotoxic markers (rare+, granzyme, perforin, TIA1)
__________________________________________
DDx: Hodgkin lymphoma (may have CD30+, CD15+ cells)
Lennert Lymphoma
Diffuse interfollicular growth c lots of small cells c nuclear irregularities, clusters of epithelioid histiocytes , large proliferating blasts, possible RS-like cells (that are EBV+)
IHC: (+) CD8

Lennert Lymphoma

Lennert Lymphoma
Lymphoepithelioid variant of PTCL, NOS. Formely known as Lennert lymphoma. Clusters of epithelioid histiocytes with atypical lymphoid cells in the intervening stroma, cells mainly small to medium, with only occasional larger immunoblast-like cells

PTCL can be CD30+, CD15+. Here it doesn't look like Hodgkin by H&E, but the IHC does

Another case of PTCL with large atypical cells mimicking the RS cells of Hodgkin lymphoma, the H&E here has a mixed inflammatory background, which may look even more worrisome
PTCL, NOS. Shows evidence of inflammatory background, scattered eosinophils and plasma cells, other cells show range in cell size.

PTCL that looks like the Reed-Sternberg (RS)-like cells of Hodgkins

PTCL NOS showing marked nuclear pleomorphism
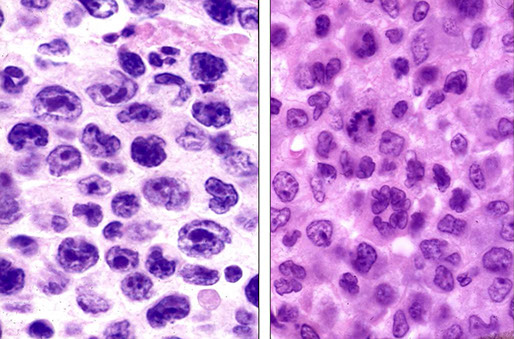
PTCL expressing CD3
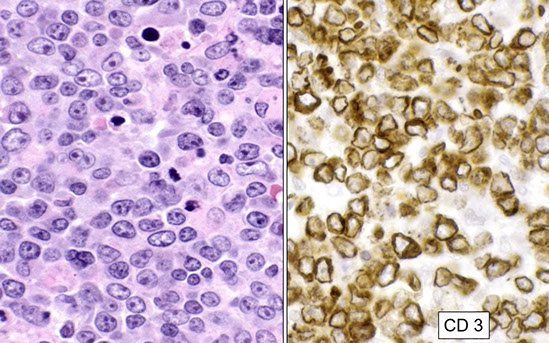
Subclassification of PTCL NOS by gene expression profiling. One subgroup has Th1 pattern with TBX21/TBET expression and the other the Th2 pattern with GATA3 expression, some cases were unclassifiable

Subclassification of PTCL may effect prognosis, GATA3 worst
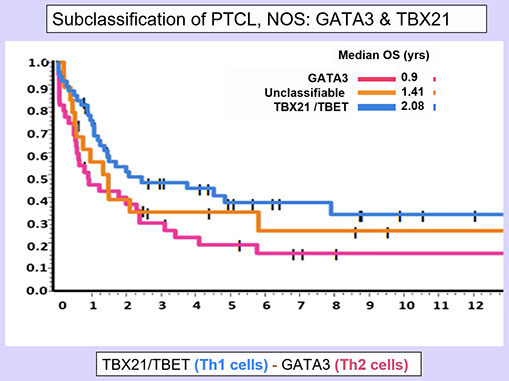
Angioimmunoblastic T-cell lymphoma and other nodal lymphomas of T follicular helper (TFH) cell origin
Gene expression profiling has helped to categorize PTCLs

PTCL with EBV+ Hodgkin/Reed-Sternberg (HRS) cells, a pitfall in the diagnosis of AITL, looks like Hodgkin lymphoma

Angioimmunoblastic T-cell lymphoma (AITL)
One of the MC peripheral T cell lymphomas, presents abruptly c with systemic dz (rash, cyroglobulins or cold agglutinins [Coombs+], hypergammaglobulinemia, RF abs, ASMA abs)
- an infiltrate into LNs of pleomorphic cells esp in the high endothelial venules and follicular dendritic cells
- occurs a little later in life, M=F, commonly involves skin and BM in addition to LNs
- has EBV assoc, although neoplastic T-cells are EBV-negative
- pts become immunodeficient 2/2 the cancer
- derived from CD4+ follicular helper T-cell (usually in follicular germinal center helping to educate B-cells) which stimulate the high-endothelial venules causing them to look big and nasty
Originally thought to be an abnormal reactive process, a disorder of immune regulation, later defined as a form of PTCL
- the clinical syndrome is a nearly universal part of the disease definition
- one would be hesitant to make the diagnosis in the absence of the characteristic clinical picture
Clinical features:
Older adults, generalized lymphadenopathy, hepatosplenomegaly, skin rash, effusions, fever
Polyclonal hypergammaglobulinemia, hemolytic anemia
Diverse constitutional signs and symptoms
AITL may be a "stem cell" disease, since the earliest mutations in TET2 can occur in B-cells and T-cells
__________________________________________
Labs: circulating immune complexes, cold agglutinins c hemolytic anemia, (+) Rf factor and anti-SM abs
__________________________________________
Micro: arborizing vasculature, atypical lymphocytes +/- clear cytoplasm, scattered B-immunoblasts, plasmacytiosis, eosinophils, regressed or absent follicles in most cases, some cases with follicular hyperplasia (type 1 pattern)
- neoplastic T-cells often with increased clear cytoplasm
- dilated subcapsular sinuses with extension of the neoplasm beyond the sinuses
Partial effacement of node, in postcapillary venules see immunoblasts, lymph, plasma cells (may be abundant), eos and aggs of cells c clear cytoplasm; perinodal infiltration c sparing of peripheral cortical sinuses
- expansion of B immunoblasts is usually present in paracortex
- can find EBV-(+) Hodgkin/Reed-Sternberg (HRS)-like cells
- follicular dendritic cell entrapping of HEV
__________________________________________
IHC: (+) T-cell antigens (CD4+), PD1+, CD10+, ICOS+, BCL6 +/- T-cells, CXCL 13 and PD1 surrounding HEV
- CD21/23 highlights expanded dendritic cell networks around HEVs
- EBV+ (in 80-90%); scattered large B-cells, T-cells are negative for EBV
- polyclonal, rarely monoclonal plasma cells
- loss of surface CD3 is very commonly seen, in cells which are also CD10+ and CD4+
- neg: can have loss of CD5/7/8
__________________________________________
Genes: usually (>90%) has clonal gene rearrangements of T-cell receptor, 10-30% with IG cloncally arranged
- mutations in TET2, IDH2, DNMT3A, RHOA, CD28, there is someoverlap between AITL and other PTCLs
-- TET2 is an early event in the pathogenesis of AITL
- Gene fusions ITK-SYK, CTLA4-CD28
- IDH2 most specific for AITL
__________________________________________
DDx: EBV+ DLBCL, Classic Hodgkin Lymphoma (cHL)
__________________________________________
Px: aggressive (<3 years survival)
- although can see clonal proliferation of B-cells (even DLBCL) or RS-like cells, best to just diagnose as AITL since the prognosis is most dependent on the poorly-performing AITL and the B-cell proliferaitons are less clinically significant [16]

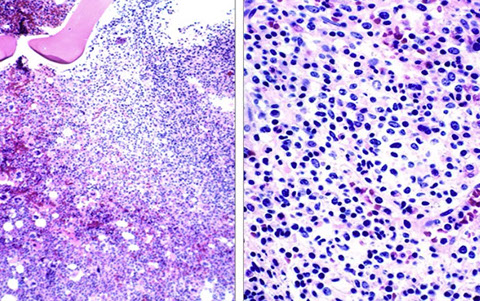

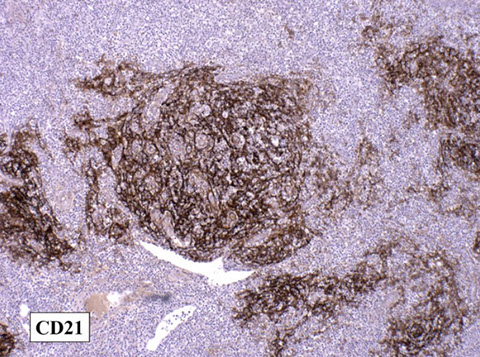

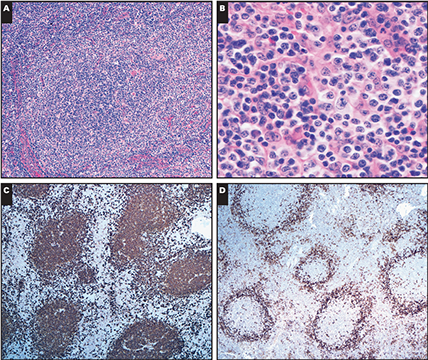
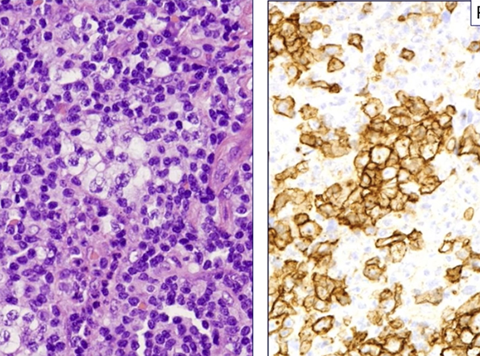
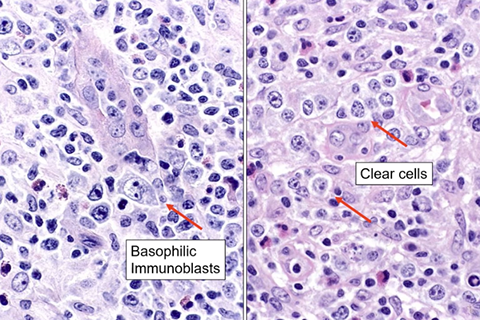


AITL. Helpful feature is the dilatation of the peripheral cortical sinus. Infiltrate extends beyond the sinus into the adjacent nodal capsule.
AITL in BM. Intertrabecular infiltrates. Loose lymphoid nodules.
AITL. CD20. B-cell areas usually reduced, seen peripheralized here to the cortex.
AITL, CD21 highlighting FDC meshwork around HEVs. Should also look at B-cell stain, and examine that these are not B-cells in the meshwork, but around the HEVs
EBV+ cells in AITL
AITL with plump HEVs and monocytoid cells [20]
AITL, low-power [20]
Angioimmunoblastic T-cell lymphoma pattern I. A, Reactive germinal center is surrounded by atypical cells with clear cytoplasm and immunoblastic features. High endothelial venules surround the atypical follicular structure. B, The perifollicular cells have clear cytoplasm; admixed plasma cells are present. C, CD20 highlights the germinal centers and scattered interfollicular B cells. D, PD1 is strongly positive in the perifollicular T cells. [16]
PD1 expression in AITL. Must use caution with PD1 because PD1+ T-cells are invariably present in reactive paracortical hyperplasia!!!!!
AITL. Higher power, arborizing vasculature
AITL. Polymorphous infiltrate. Scattered large cells, being both clear cells (the atypical T-cells) on the right, and immnuoblasts that are B-cell derived on the left. Note scattered eos and epithelioid histiocytes.
CD10+ in AITL, which is slightly less than CD3
1 - 12
<
>
Comparison of patterns in AITL [16]

Follicular T-cell lymphoma
- aka the follicular variant of PTCL (??)
Lymph node based, TFH cells with predominantly follicular growth pattern
- round nuclei, abundant pale cytoplasm
Intrafolicular T-cell lymphomas derived from TFH cells
- grouped with AITL in revised WHO classification, although lacks characteristic morphologic features of AITL (eg HEV proliferation, extrafollicular dendritic meshwork proliferation)
- clinical syndrome otherwise similar to AITL (generalized LAD, splenomegaly, B-sx, skin rash)
- AITL may be seen in subsequent biopsies
Micro: Clusters of clear cells within follicles
- may simulate follicular lymphoma
- can have second morphologic pattern mimicking progressive transformation of germinal centers (PTGC)
IHC: Usually CD4+, BCL6+, ICOS+, CD10 +/-, PD1/CD279+
- may also have EBV+, RS-like cells
Genes: IYT-SYK fusion in 20% of cases, appears to be fairly specific
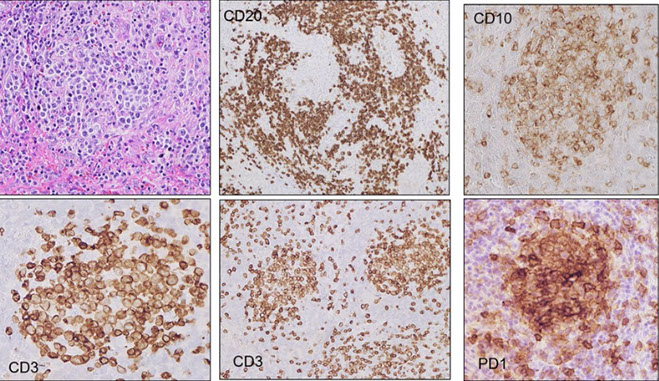






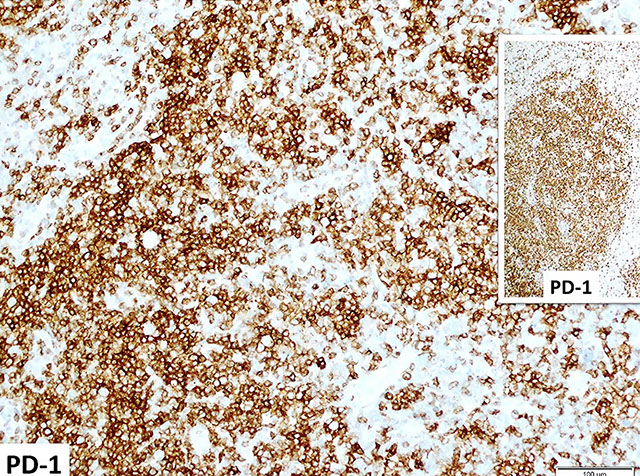
Follicular T-cell lymphoma. (clockwise from upper left) H&E shows vaguely nodular growth pattern. CD20 shows some holes in the follicles, which are filled with T-cells expressing CD10, PD1, CD3
Pronounced nodular lymphoid proliferation extending into adjacent soft tissue [22]
Monomorphic proliferation of medium-sized cells with a bit of clear cytoplasm [22]
Also monomorphic population with some clear cytoplasm.
Mostly T-cells with some interstpersed B-cells
Mostly CD4+ cells
T-cells express BCL6 and CD10
Absence of dendritic meshworks, CD23 showed a similar pattern
PD1 stained cells in a follicular pattern
1 - 9
<
>
Nodal peripheral T-cell lymphoma with TFH phenotype
- Subset of PTCL with TFH phenotype, as well as some of the features of AITL (may represent a tumor-cell rich variant of AITL)
- should have at least 2 TFH-cell markers in addition to CD4
IHC: (+) CD4, PD1, CD10, BCL6, CXCL13, ICOS
Genes: shares some of the genetic alterations seen in AITL, such as mutations of TET2, DNMT3, and RHOA
Anaplastic large cell lymphoma (ALCL), ALK positive
T cell lymphoma MC in (slightly) males up to 30 yo (younger pts than ALK neg, usually children and young adults), 1/2 of HG-lymphomas of childhood
- MC in non-contiguous inguinal LNs, 7/10 presenting with advanced stage disease
- assoc c pulmonary inflam pseudotumors, HIV, MF
- may have skin manifestation as the sole manifestation of the disease (vs pcALCL?), commonly involves extanodal sites (skin, bone, soft tissue)
- 5 patterns: common (60%), lymphohistiocytic (10%), small cell (5-10%), Hodgkin-like (3%), composite pattern (15%)
Common pattern (60%)
sheets or clusters of hallmark cells that efface LN architecture
- tumor cells often with sinusoidal growth pattern, where tumor cells expand sinuses and can mimic metastatic tumors
- cells with multiple nuclei arranged in a wreath-like pattern can be seen
Lymphohistiocytic (10%) or Histiocyte-Rich variant
- Common in children and young adults
- Abundant histiocytes with pink cytoplasm mask minor population of tumor cells
- May be mistaken for infectious or reactive process - delay in diagnosis is common
- Recurrent biopsies contain fewer histiocytes, more tumor cells
Small cell variant (5-10%)
- common in children and young adults
- usually presents in LNs
- most tumor cells are small with clear cytoplasm, irregular nuclei
- minor population of classical ALCL cells, often around blood vessels
- may be mistaken for PTCL, NOS, but neoplastic cells are often CD3 negative
- ALK+, but small cells may show variable staining
___________________________________________
Micro: anaplastic cells c lots of cytoplasm clustering around BVs, horse-shoe nuclei (hallmark cells), and ALK (anaplastic lymphoma kinase) transcription
- sinusoidal growth pattern
- perivesicular rosetting (does not destroy BVs)
- small cell variant made of large lymphs which are not anaplstic (which can be confused c PTCL)
The ultimate histologic spectrum of ALCL is both broader and narrower than the original concept [12]
- small cell and histiocytic variants included
- most Hodgkin-like and highly pleomorphic CD30+ lymphomas excluded
- new diagnostic tools enhance our diagnostic acumen on routne H&E (eg "Hallmark cells")
- Importantly, what we recognize as ALCL has cliical and prognostic significance [12]
Histologic spectrum of ALK+ ALCL [12]
- marked variation in cell size from case to case
--- hallmark cells are always present
- distinctive architectural features
--- sinusoidal infiltration
--- perivascular rosetting
- Cellular density varies markedly
--- hypercellular to hypocellular
--- hypocellular cases may simulate an inflammatory process
___________________________________________
IHC: (+) CD30 (membranous and golgi), cytotoxic proteins (granzyme+, TIA-1+, CD25+), ALK+ (cytoplasmic and nuclear with the t(2;5), clusterin+, variable CD45 / 45RO / 43, some T-cell markers, EMA (var), PAX5 (rarely)
- CD3, CD5, and CD7 often lost; CD2 and CD43 often positive
- neg: CD15, EBV, B-cell ags, CD3 often negative!
- CD30 can be lost with anti-CD30 therapy
May have "null-cell"phenotype, where only evidence of T-cell differentiation is at a genetic level
Note: Primary cutaneous ALCL usually lacks expression of EMA and ALK; if when Nucleophosmin (NPM):ALK detected as a nuclear/cytoplasmic stain, a thorough investigation should be done to exclude nodal systemic ALCL
___________________________________________
Genes: ALK expression assoc c t(2;5) NPM gene mutations, usually NPM/ALK in cytoplasm and nucleus, seen in 84%
- t(1;2) assoc c TPM3 gene mutations, membranous and cytoplasmic Alk staining, seen in 13%
- t(2;3) assoc c TFG gene mutations, cytoplasmic Alk staining
Don't need to do FISH for ALK break apart proteins if the ALK IHC is positive
___________________________________________
DDx: Hodgkin lymphoma (has + B cell markers); LyP, ALK-positive DLBCL, pcALCL, large cell transformation fo MF
- ALK+ histiocytosis (occurs in infancy)
___________________________________________
Tx: chemo, should excise skin lesions
- currently lookin at ALK inhibition
___________________________________________
Px: better than other PTCLs, and better than ALK neg (although the difference with ALK neg may be 2/2 age)
- the small cell and histiocytic variants have a worse px because usually disseminated at dx

ALCL, CD30
ALCL


ALCL - ALK+


(A) ALK2 ALCL with DUSP22 rearrangement.
There is a relatively monotonous proliferation of
large transformed cells and classic “Hallmark” cells.

(B) Breast implant–associated ALCL. The seroma cavity demonstrates numerous very large anaplastic-appearing lymphoid cells.
ALCL hallmark cell

ALCL with a myxoid stroma



ALK-positive anaplastic large cell lymphoma (ALCL), small cell variant (A)showing diffuse infiltrate small to medium-sized cells with irregular nuclei effacing the nodal architecture. Many of the cells have pale cytoplasm and a centrally located nucleus (fried egg appearance). Rare hallmark cells with reniform nuclei are seen (B) ALK staining shows a nuclear and cytoplasmic staining pattern[15]
Evolution of ALCL [12]

Small cell variant of ALCL
ALCL. ALK and CD30 highlight classic sinusoidal infiltration by neoplastic cells

ALCL is not characterized by highly pleomorphic cells!! See perivascular rosetting (on the right) do not see necrosis or vacsular blockage though

ALK staining can vary based on the translocation present

Lymphohistiocytic ALCL. Right is CD30
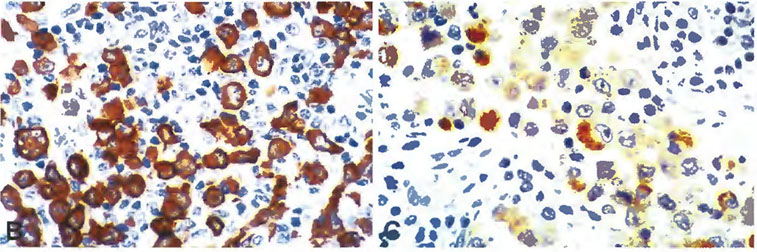
Different ALK staining patterns in ALCL by translocation. (B) Membranous and diffuse cytoplasmic with t(1;2). (C) granular cytoplasic with t(2;17). Not shown is nuclear and diffuse cytoplasmic with t(2;5)
Anaplastic large cell lymphoma, ALK negative
Lymphoma histologically resembling ALK(+) ALCL with positive CD30, but lacking ALK expression
- Rarely develops in patients with silicone breast implants
__________________________________________
Micro: Hallmark cells less frequent in ALK- ALCL than ALK+ ALCL
- ALK- ALCL more anaplastic than ALK(+) ALCL
__________________________________________
IHC: BCL2 , CD15 and EBER can be (+) in ALK(-) ALCL, but are (-) in ALK(+) ALCL
Need a cohesive growth pattern of hallmark-like cells with strong and uniform CD30 expression (usually Golgi and membranous)
- EMA+, cytotoxic+, sinusoidal growth, and loss of T-cell ag are desirable but not essential
__________________________________________
Genes: ALK(+) and ALK(-) ALCL have different gene expression profiles confirms they are different dz's
- DUSP22 rearrangements and IRF4 rearrangements:
cr 6p25, detected in ~ 30% of ALK(-) ALCL cases, better prognosis
- DUSP22 mutated ALCL have typical hallmark cells and are negative for Granzyme B (and other cytotoxic markers)
– TP63 rearrangements reported in ~ 8% of ALK(-) ALCL cases, worse prognosis
DUSP22 translocations also seen in primary cutaneous CD30+ T LPD
- there is some evidence for a close relationship between some cases of systemic and cutaneous ALCL
__________________________________________
DDx: distinction c CD30+ PTCL NOS can be difficult
- if in the skin and ALK-negative, should consider Lymphomatoid Papulosis (LyP), Primary Cutaneous anaplastic large cell lymphoma (PC-ALCL), systemic/nodal ALCL, and CD30+ large cell transformation of mycosis fungoides (MF)
- clinical workup is essential to sort this out
__________________________________________
Px: poorer than ALK+ ALCL, but worse than PTCL-U
- subset of cases with DUSP22 rearrangement have a px similar to ALCL ALK+ [Feldman et al. Blood 2014], p63 mutated cases do a lot worse
- patients lacking rearranements of ALK, DUSP22, and TP63 (triple negative) have an intermediated px
ALCL, ALK negative. Similar morphology as ALCL ALK+. Red arrows are hallmark cells

DUSP22 mutated ALCL has a distinct morphology and phenotype

Requirements for ALCL ALK neg.

Hodgkin's-like ALCL is NOT ALCL, but usually a B-cell derived neoplasm, such as aggressive for of Hodgkin dz
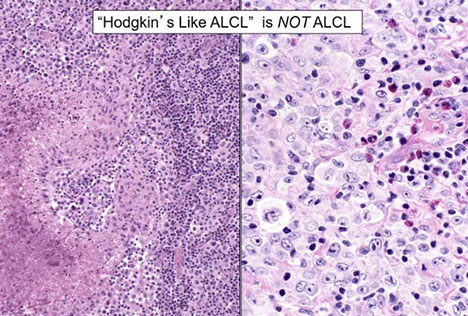
Breast implant-associated anaplastic large cell lymphoma
- aka BI-ALCL, or BIA-ALCL
Provisional entity; morphologically and immunohistochemically the same as ALK-negative ALCL
- presents as accumulation of seroma fluid bwt implant and surrounding fibrous capsule
-- diagnosis is best made by cytology
- ~10 yr interval from time of implant to lymphoma (though the timeframe can vary greatly)
- neoplastic cells usually confined to seroma fluid and do not invade capsule
~30% have axillary lymphadenopathy
- highest risk with textured impoants [12]
- may arise from an activated mature cytotoxic T-cell
Clonal TCR reported in most but not all cases
- JAK/STAT pathway mutations in 59% (STAT3/JAK1 most common)
- epigenetic modifiers mutated in 74%
__________________________________________
Micro: tumor cells lines the capsule and may infiltrate the capsule
- tumor cells are large and pleomorphic, usually can see the hallmark cells characteristic of ALCL
__________________________________________
IHC: similar to ALK-negative ALCL; tumor cells CD30+, negative for ALK; may show loss of T-cell antigens
__________________________________________
Genes: TR genes clonally rearranged in most cases
- may have complex karyotypes
- Recurrent activating JAK1 and STAT3 mutations have been reported
- does not have DUSP22 or TP63 mutations
__________________________________________
Ddx: carcinomas, angiosarcomas, other lymphomas
__________________________________________
Tx: removal of implant is probably adequate therapy in most cases - if no invasion of capsule [12]
__________________________________________
Px: most patients have an excellent outcome
- median survival is 12 years
- most important prognostic factor is the presence of a solid mass of tumor cells, which may mean systemic therapy is needed
93% CR in pts with disease confined to capsule
72% CR in patients with a mass
No difference in OS or PFS in patients who had chemorx (at least with local disease)
BIA-ALCL anatomy [12]

BIA-ALCL seroma cytology. Typical cells with deeply basophilic cytoplasm and lobulated nuclear contours

BIA-ALCL capsule. May be only rare atypical cells.
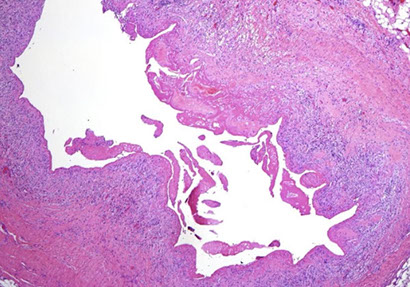
BIA-ALCL IHC. CD30 lining seroma cavity.

BIA-ALCL capsule high power, without obvious invasion

Hodgkin Lymphoma
Overview
All have:
- origin in lymph nodes, esp in cervical region
- occur in young adults
- neoplasm has scattered Reed-Sternberg (RS) cells amidst non-neoplastic inflam cells
- often surrounded by T-cells
1832: T. Hodgkin wrote his paper: "On some morbid appearances of the absorbent glands and spleen", comprising seven cases, including the case of Carswell
- In 1865, S. Wilks reports 15 "Cases of enlargement of the lymphativ glands and spleen", where he proposes the eponym "Hodgkin's disease"
What happened to Hodgkin the person?
1837 - T. Hodgkin fails to get post as Asst. Physician at Guy's Hospital (he was a Quaker and social activist -- had crossed the administration)
1857 -- Travels to the Holy Land with Sir Moses Montefiore
1866 - Died in Jaffa during a choleric epidemic while distributing relief to local Jewish population
1902 - D. Reed described features of neoplastic cells (Reed-Sternberg cells) that hold true today
- she disputed view of Sternberg, who proposed in 1898 that Hodgkin's lymphoma was a chronic inflammatory process related to tuberculosis
Classic paper by D. Reed: On the Pathological Changes in Hodgkin's Disease with Especial Reference to its Relation to Tuberculosis. DM Reed, JHH Reports 10: 133-199, 1902
- her 1902 paper noted that "the general health of the patient before the onset of the disease is usually excellent"
- noted the early peak among children and young adults
- noted high incidence of anergy
- prsents as painless progressive cervical adenopathy without leukemia
- proposes that the pathological picture is sufficient for diagnosis
- Dorothy Reed graduated in 1900, 5th in her class
- Did internship in medicine under Osler, the a pathology fellowship
- published a case of lymphatic leukemia, proposing an origin from the bone marrow, in addition to one other paper during her pathology fellowship
- Seeks post as an "assistant" in pathology from Chairman WM. Welch, which Welch denied because she was a woman
Hodgkin's diisease vs Hodgkin's lymphoma
- the name "Hodgkin's disease" was based on the uncertain status of HD, infectious or neoplastic?
- the neoplastic nature is proven
- the cells of origin of HD was uncertain -- histiocyte, reticulum cells, lymphocyte, granulocyte?? --- A B-lymphocyte origin is now proven for nearly all cases
- Therefore, the name "Hodgkin's lymphoma" reflects our modern understanding
Comprise ~30% of all lymphomas
- vs non-Hodgkin lymphoma, incidence has remained constant (not increasing)
Hodgkin's lymphoma (WHO)
Classical Hodgkin's Lymphoma
- Nodular sclerosis (70%) --- grade 1-2 (optional)
- Mixed cellularity (10-15%)
- Lymphocyte depleted (<5%)
CHL, lymphocyte-rich (5%) -- nodular and diffuse
Nodular lymphocyte predominant Hodgkin's lymphoma
Named by Thomas Hodgkin in early 1800's
- RS cell is of B-cell origin, thus Hodgkin lymphoma (not Hodgkin disease)
Broken down into 2 entities: 1) classical Hodgkin lymphoma (CHL) and 2) Nodular Lymphocyte Predominant Hodgkin Lymphoma (NLPHL) [discussed below]
- differ in clinical features, behavior, morphology, IHC, B-cell survival
Classic Hodgkin's Lymphoma: Genetic features
- Single cell PCR:
---- Ig genes rearranged, clonal, somatic mutation of VH genes
---- No Ig production - many theories: Crippling mutations, Defects in IGH transcription factors, Methylation / silencing of IG genes
- Loss of the B-cell program
--- most B-cell markers negative, except PAX5
Diseases that can have HRS-like cells:
- EBV+ Mucocutaneous Ulcer
- B-cell lymphoma, unclassifiable with features intermediate between DLBCL and cHL
- ALCL
- ATLL
- AITL
- PTCL
- cHL
- cHL PTLD
- NLPHL
- T/HRLBCL
- CLL transformed to cHL
- EBV+ DLBCL
- PMBL
First case of Hodgkin lymphoma published in 1828 by Robert Carswell as "Cancer cerebriformis of the lymphatic glands and spleen"

Case 2 of Hodgkin disease by T. Hodgkin, Guy's Hospital (London). with splenic and LN involvement

RS cells -- by Dorothy Reed

Image from the original paper by DM Reed at JH

Hodgkin Lymphoma comes from B-cells [10]

Classic immunophenotype CHL - positive for CD30 and CD15, rosettes with CD3, and variable positivity for CD20

Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL)
WHO 2016 definition:
- B-cell neoplasm c nodular / nodular-diffuse prolif of scattered large neoplastic cells called popcorn or Lymphocyte predominant (LP) cells
- LP cells ringed by PD1/CD279+ T-cells
- In typical cases, LP cells reside in large spherical meshworks of follicular dendritic cell processes that are filled with non-neoplastic lymphocytes (mainly B-cells) and histiocytes
~5% of Hodgkin lymphomas (not too common...), in young adults (4th-5th decades), children, M>F
Most are localized (stage I-II) >80%, peripheral nodes (cervical, axillary, inguinal), spares the mediastinum, axial LNs, and BM [10]
Course: Indolent, long natural history, >95% survival
- late (multiple) relapses more common; localized, not fatal
- association with or progression to LBCL (THRLBCL in 2-3%)
B-cell neoplasm c nodular / nodular-diffuse prolif of scattered large neoplastic cells called popcorn or Lymphocyte predominant (LP) cells
- formerly known as Lymphocytic and Histiocytic (L&H) RS cell var
- classic RS cell sparse to absent
- progressive transformation to GC (PTGC) is NOT a precursor lesion, most PTGCs do not go to NLPHL
5% of all HLs
- usually in LNs, but BM, mediastinal and splenic involvement rare
- 1 nodule c features of NLPHL in diffuse growth pattern excludes dx of THRLBCL
Associated with Progressive Transformation of Germinal Centers (PTGC)
- can involve the same LN
Micro: prolif of B cells c few background has CD3 / PD1+ T cells encircling neoplastic LP cells (rosettes), CD21 shows residual follicular network, neoplastic cells have CD20/EMA+
Cells found in spherical networks of follicular / nodular dendritic cell networks full of non-neoplastic lymphs and histiocytes
- 1/3 of NLPHL dx'd in past are actually lymph-rich CHL
-- a pure form of NLPHL may not even exist...
Nodular / diffuse infiltrate of small lymphocytes, histiocytes, and LP cells (large folded uninucleus, scanty cytoplasm [popcorn cells, due to lots of nuclear folding]) replaces LN architecture
- may be difficult to distinguish LP from RS cells
- diffuse areas have small lymphs c histiocytes
- histiocytes and plasma cells can be seen at margin of nodules
LP cells: (+) CD20 (strong)/45/75/79a, BCL6, EMA (50%), sIgD (subset, up to 25%), OCT-2 and BOB.1 strongly expressed
(+) AID, PAX5 (strong)
(-) CD15/30 usually negative (though can see CD30 [+] large cells/LP cells), fascin, EBV
- LP cells (formerly L&H cells) cells rosetted by Follicular Tfh cells, that are PD-1+, CD57+
- PD-L1 usually negative
- rarely can have EBV+ cases of NLPHL, and EBV causes upregulation of CD30 --- may look like CHL!!
CD45 is by definition always negative in classic Hodgkin Lymphoma (cHL),
- background has subset of germinal center T-cells which are (+) CD4/57/134, c-MAF, PD-1, BCL6, IFR4/MUM1
-- (-) TIA1, CD40L
- lots of these T-cells in diffuse areas, and inc over time
IgD+ NLPHL in younger pts, M>>F (23:1), cervical LNs, GC: CD38wk+ IgM- CD27-, PU.1 (a B-cell marker) staining in 60% of IgD, extrafollicular LP cells are more often positive, may be more predictive of THRLBCL (19/20 IgD+)
CD21 essential for recognizing a follicular dendritic cell pattern
Good to distinguish NLPHL that progress diffuse T-cell rich pattern as NLPHL, THRLBCL-like from primary THRLBCL
Absence of CD+/TIA1+ cells favors THRLBCL
Genetic rearrangements present in single LP cells, c IG VH genes carrying high rates of somatic mutations
- EBV absent in LP cells, but present in nearby lymphs
DDx:
CHL (CD20+/33%, CD30+/97%, CD15+/85%) vs NLPHL (CD20+ in 98%, CD30 and CD15+ in 5%)
- other useful markers are CD21/CD23 for the follicular dendritic network, PD-1 for T-cell rosetting, CD45+ and EBV in NLPHL
- B-cell transcription factors (PAX5, OCT2, and BOB1) are downregulated in CHL and nicely + in NLPHL
- NLPHL is strongly + for other B-cell markers (CD79a) and expresses Kappa or Lambda
Px" Slowly developing dz, c frequent relapses
- rarely fatal; responsive to tx
Stage I and II have esp good px (10yr survival >80%)
- advanced stages have less favorable px
- 3-5% of cases progress to large B-cell lymphoma
- BM involvement is rare
Histologic Progression in NLPHL [10]
DLBCL in 5% of NLPHL; clonally related by Ig gene PCR
May evolve to a T-cell histiocyte-rich LBCL (THRBLCL), which is a completely diffuse T-cell-rich proliferation lacking any follicular dendritic cells
- PD1 is useful in distinguishing NLPHL from THRLBCL bc shows rosetted in NLPHL which are absent in THRLBCL (although, PD1+ rosettes are not unique to NLPHL, can also be seen in PTGC, EBV, HIV, and RDD)
- controversial if these cases differ from de novo T/HRLBCL
- common genetic aberrations in NLPHL/T/HRLBCL (JUNB, DUSP2, SGK1, SOCS1, and CREBBP
- probably a continuous disease spectrum
- cases with progression are diagnosed as "TC/HRLBCL-like transformation of NLPHL" per the WHO 2017
- stage is an important progrnostic factor in the clinical management in NLPHL, with or without progression
- this is assoc c an aggressive clinical course
NLPHL. low pwer, with large nodules that often have epithelioid histiocytes, and small epithelioid granulomas at the periphery of the nodules, with absence of fibrous bands that you would see in Hodgkin's,

NLPHL popcorn cells


Many of the atypical cells had multilobulated nuclei and others had large, rounded nuclei with multiple nucleoli. The large cells were very tightly ringed by smaller lymphocytes forming rosettes (panel A; original magnification ×1000, hematoxylin and eosin stain). The large cells and the majority of small background lymphocytes were CD20 positive (arrows, panel B; original magnification ×1000, CD20 immunostain). However, the small lymphocytes ringing the large cells were distinctly CD20 negative (panel B). These lymphocytes expressed CD3 (arrow, panel C; original magnification ×1000, CD3 immunostain), programmed cell death protein-1 (arrows, panel D; original magnification ×1000, PD-1 immunostain), and CD57, consistent with follicular helper T (TFH) cells. Additional studies showed that the tumor cells expressed PAX5 and BCL6 but were negative for CD30, CD15, and Epstein-Barr virus. A diagnosis of nodular lymphocyte–predominant Hodgkin lymphoma (NLPHL) was made.
IHC of NLPHL. neoplastic cells uniformly CD20+, with relatively frequent small CD20+ cells in the background, the large neopalstic cells are rosetted by T-cells

OCT2 is an important marker in the diagnosis of NLPHL, strongly stains the LP cells, highlighting the nuclear atypia, with the irregular nuclear contour, with a staining that is more intense the the background of small B-cells. Each of the neoplastic LP cells is surrounded by a clear area composed of rosetting T-cells

NLPHL follicular dendritic cell network

NLPHL. 6 main patterns that are incorporated in the WHO. Pattern A: "Classical" B-cell rich nodular. Pattern B: Serpiginous / Interconnected. Pattern C: Prominent extra-Nodular LH cells. Pattern D: T-cell rich nodular. Pattern E: Diffuse (TCRBCL or DLBCL-like), predominance of pattern E is indicative of recurrence!!, and Pattern F: Diffuse moth-eaten, B-cell rich

NLPHL progression. Nodular to diffuse architecture with loss of FDC meshworks, B-cell rich to T-cell rich background. NLPHL with THRBCL areas (pattern E) shows overlap with THRLBCL and this distinction may be difficult if not impossible in small biopsies

NLPHL. CD3 stain highlighting the numerous reactive T-cells, which may mask LP cells and lead to the erroneous diagnosis of a T-cell lymphoma

NLPHL unexpectantly IgD+, background B-cells positive for IgD, but the neoplastic LP cells rosetted by CD3+ cells. IgD+ has clinical implications
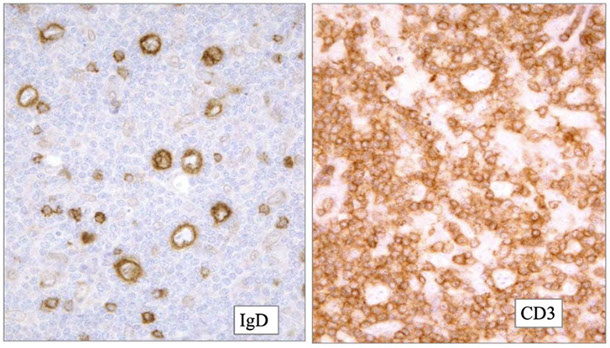

Significance of IgD in NLPHL
- IgD expression also associated with Moraxella catarrhalis in young boys (Hartmann et al 2015)
NLPHL with progression to DLBCL, with the top images showing NLPHL with scattered LP cells, and the bottom showing sheets of neoplstic cells more typical of DLBCL that are positive with CD20

Differentiating NLPHL from T/HRLBCL
NLPHL with progression to T/HRLBCL. Upper panel with axillary LN diagnosed as NLPHL and shows typical features with scattered LP cells and the presence of small B-cell in background and nodular growth pattern. But the BM showed features of T/HRLBCL indicating stage 4 disease, mandating treatment as DLBCL
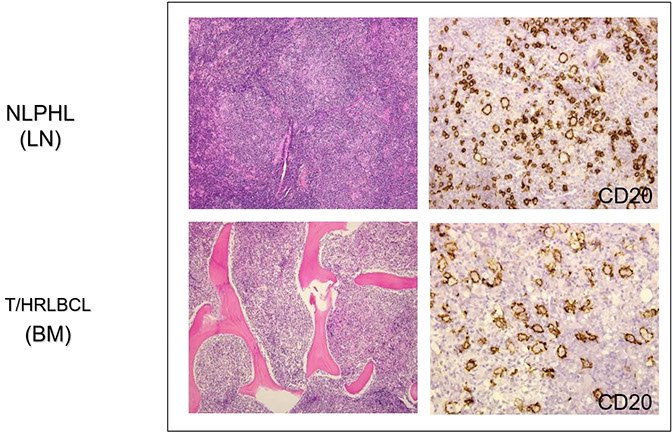
classical Hodgkin Lymphoma (cHL)
NSCHL (70%) > MCHL (20%) > NLPHL = LRCHL (4-5% each) > LDCHL (>1%)
CHL has 4 subtypes: 1) Nodular sclerosis, 2) mixed cellularity, 3) lymphocyte-rich, 4) lymphocyte-depleted
- differ in sites of involvement, clinical presentation, amt of fibrosis, cellular composition, amt of atypia, presence of EBV infx
- incidence bimodal: at 15-30 yo and after 50 yo
- most have localized lymphadenopathy (usually cervical nodes or mediastinum) spreading by contiguous lymph sites; may have "B" sx
- Reed-Sternberg (RS) cells have the binucleated owl's eye appearance, whereas Hodgkin's cells are mononuclear
CD45 is by definition always negative in RS cells in cHL [8]
Aberrant T-cell ag expression in CHL [10]
- aberrant expression of CD2, CD4, CD3 was in 5% of CHL
- HRS cells expressing T-ag often higher grade (CHL, NS2)
- in most cases, HRS cells are genotypically G-cell (IGHR+) and lack TCR rearrangement
- PAX5 is still positive, indicative of B-cell lineage
- Misdiagnosis as Anaplastic large cell lymphoma, ALK-negative is a common pitfall
DDx: EBV+ B-cell blasts may assume HRS-cell morphology and immunophenotype, and are seen in many EBV+ B-cell lymphoproliferations
- Infectious mononucleosis, Iatrogenic LPD (eg Methotrexate), Age-related EBV+ B-cell lymphomas, PTLD
EBV+ HRS-like cells described in AILT, ATLL, and PTL-NOS
EBV+ B-cells in PTCL may have HRS like features - most common in AITL, other nodal PTL (NOS, ATLL)
- some PTCL have a Hodgkin's lie phenotype (CD30+, CD15+),
- some cases of cHL (esp the nodular sclerosing (NS) type) may show aberrant T-ag expression and are associated with a worse prognosis

Immunophenotype of a classic Hodgkin lymphoma. See variable expression of CD20 in the RS cells, the RS cells are surrounded by reactive T-cells
CHL DDx: May think it is a T-cell lymphoma because of aberrant T-cell marker expression, such as in the nodular sclerosing (NS) NSCHL seen here expressing CD3, CD4, and CD5, with dim expression of CD20
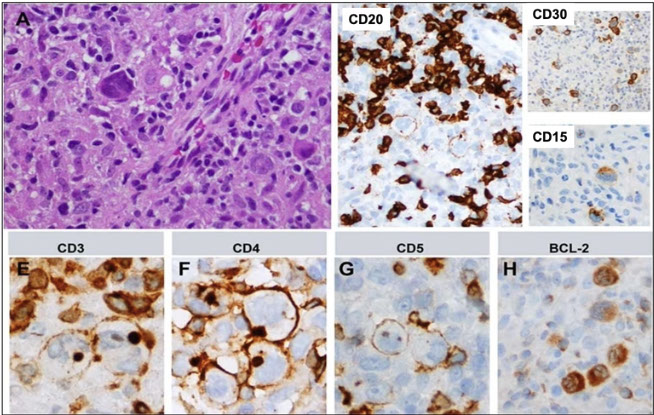
DDx: May think CHL is a T-cell lymphoma, and vice versa
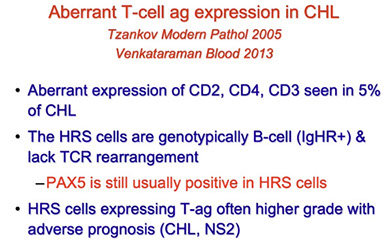
DDx: NSCHL (left) and PMBL (right).

The most common T-antigens expressed by cHL, CD4 is the most common, CD2 and CD3 less common)

Survival is worse for cHL that express T-cell antigens

DDx of Hodgkin's Lymphoma

1) Nodular sclerosis Hodgkin Lymphoma (NSHL, NSCHL)
MC type (7/10) of cHL, usually mediastinal; Nodular LN architecture separated by bands of fibrosis
- present in the second to third decase, peak incidence, F>M, stage II with mediastinal mass and supraclavicular lymphadenopathy
Lacunar (H) cells (fixation/retraction artifact) aggregate in the center of nodules are artifacts caused by formalin fixation; and has RS cells
- "mummified" cells are pyknotic c reddish nuclei
Grading: prognostic value for intermediate/high stage dz of German Hodgkin Lymphoma study group, includes qualifying the numbers of RS and H cells as sacattered (Grade 1) or aggregates (grade 2), and also tissue eosinophilia (>5% of cell infiltrates)
- syncytial NS is aggressive form that presents at high stage c bulky mediastinal dz made of sheets of RS cells and RS cell variants; that can have focal necrosis
- has some overlap with primary mediastinal large B-cell lymphoma (PMBL)
IHC: CD30+ in >95%, CD15+ in 70-80%, (CD15 and CD30 with characteristic membranous and Golgi staining) PAX5 dim in ~90%,
- MUM1/IRF4+, p53+
- EBV+ with latency II pattern in 20% (LMP1+, LMP2+, EBNA1+, EBNA2-)
- EBER ISH + in 20%
- fascin +/-, BCL2+/-
- BOB1 and OCT2, -/+, PU.1 -
- RS+H cells aberrantly express T-cell markers in 15%
- Background CD4+ rosettes around RS+H cells
- increased histiocytes correlated with poorer px
- CD45 always negative in cHL
Px: >90% 5yr survival in pts c early disease
- Px depends on the stage, bulk of disease
- histological significance of grading less relevant with modern therapies
NSCHL in thymus, often results in a cyst formation

Radiologic appears of NSCHL showing a mediastinal mass

Gross appearance of the mediastinal mass with cellular nodules that are yellow with white sclerotic bands

NSCHL. Lacunar variants that look more like classic RS cells, others with less conspicuous nucleoli.

NSCHL. Vaguely nodular with dense fibrosis, with lacunar cells that appear to be sitting in a space left by shrinkage.

3-crop-u29753.jpg?crc=4064522007)
2-crop-u29759.jpg?crc=124110160)
Lacunar (H) cell
Syncytium of lacunar cells in cHL - NS, syncytial variant
CHL, nodular sclerosis grade 2. Shows central necrosis, palisaded by HRS cells around central necrosis, have a higher incidence of B-cymptoms (grading is considered optional)

2) Mixed cellularity
In peripheral LNs, is up to 1/4 CHLs; HIV-assoc in developing nations;
- M>F
- bimodal age range, pediatric and older adults
- often stage III, IV
- may be associated with HIV
- EBV+ in 75%
- Classical RS-cells in a mixed inflmmatory background
- fine trabecular sclerosis but no fibrous bands
Micro: has nodular pattern without bands of fibrosis bwt nodules (though can have a little bit of fibrosis w/in nodules); usually has lots of RS cells and moderate amt of monnuclear Hodgkin cells
- 3/4 assoc c EBV
cHL - mixed cellularity


Mixed cellularity Hodgkin's lymphoma

3) Lymphocyte-rich classic Hodgkin Lymphoma (LRCHL)
Formerly included in LPHD of Rye classification
- background rich in lymphocytes with few RS cells
- Growth pattern: "Nodular" or "Diffuse"
-- nodular form is more common
-- Diffuse form may be a variant of mixed cellularity, rich in small lumphocytes
- tends to be low stage, older patients
- EBV association is variable [10]
~4% of CHL cases (pretty rare);
Similar to NLPHL but has immunophenotype of CHL (CD45 is always negative in any cHL, but can be positive in NLPHL; CD15 and CD30 show membranous and golgi staining in RS+H cells); also c nodular growth pattern and some RS cells, but few eos/neuts
- see classic RS cells, mononuclear cells, Lacunar (H) cells, popcorn cells in background of lymphs
- may form T-cell (CD3+/CD57+) rosettes around RS and H cells
IHC: 50-60% have OCT1, OCT2, and BOB1 positivity in RS+H cells
- 1/2 have EBV
Px: fairly good
Lymphocyte Rich Classical Hodgkin's, Nodular -- the nodularity is striking, and there are no fibrous bands, nodule on right kinda looks like a small regressed germinal center.

regressed follicle in LRCHL- note the predominance of small lymphocytes, the neoplastic cells are in the marginal zone in the surrounding the reminants of residual follicles

LRCHL - the nodules are rich in B-cells (CD20), holes in the nodule are where the RS CD30+, CD15+, which are rosetted by CD3+ T-cells
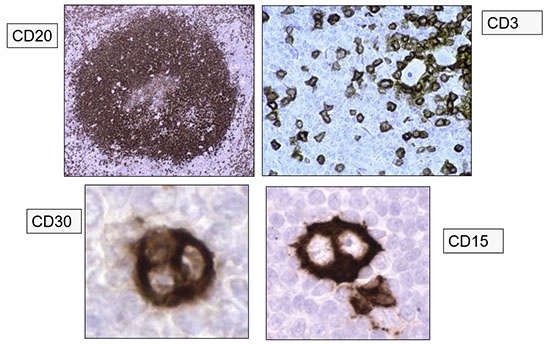
LRCHL, note the distribution of the CD15+ (left) and CD30+ (right) cells), which are in the periphery of the nodule, different from NLPHL where they are more prominent in the nodule


Hodgkin lymphoma - lymphocyte depletion, reticular subtype
Hodgkin lymphoma staging

4) Lymphocyte-depleted (LD) classic Hodgkin Lymphoma (LDCHL)
Rare (<1% of all cases of cHL) with strict criteria (many older case series were probably cases of NHL); Diffuse growth pattern that looks hypocellular 2/2 necrosis, fibrosis, little inflam;
- seen in increased frequency in developing countries and assoc c HIV in developing nations; 3/4 have EBV
- similar to mixed cellularity, but decreased normal lymphocytes, likely a continuum with MC [10]
- fibrohistiocytic background - rich in histiocytes
- sometimes called "reticular" from sheets of bizarre RS-like cells present
- has highest involvement of BM; fairly aggressive
- RS cells usually >15/hpf; also has pleomorphic cells
IHC: Reed Sternberg cells: (+) CD30, 15, IRF4/MUM1, weak Pax5, EBV (1/2+, esp MCHL, EBER), fascin
- negative B-cell markers, CD45, OCT2 and BOB.1 ([-] in 90%), EMA
-- 1/10 are CD20 weak (state in report)
-- sometimes RS cells are CD15 neg
- background lymphs are T cells
Cotswold revision of Ann Arbor staging:
I: single LN or lymph structure
II: 2+ LNs involved on same side of diaphragm (suffix indicates # involved sites)
III: Involvement on both sides of diaphragm
- III-1: +/- splenic, hilar, celiac, or portal nodes
- III-2: involved paraaortic, iliac or mesenteric nodes
IV: extranodal sites involved
Tx based on clinical features and stage of dz
ABVD regimen
Adriamycin (doxorubicin, aka Rubex) - topoisomerase inhibitor; intercalates between DNA strands
Bleomycin (aka Blenoxane) - glycopeptide antibiotic - induces DNA strand breaks
Vinblastine - anti-microtubular
Dacarbazine (DITC) - imidazole carboxamide - methylates guanine at O6 and O7
BEACOPP regimen
Bleomycin
Etoposide
Adriamycin
Cyclophosphamide
Oncovin (vincristine)
Procarbazine
Prednisone
MOPP regimen
Mustargen (chlormethine, mustine, nitrogen mustard, MSD) - alkylating agent
Oncovin (Vincristine or VCR) - binds tubulin protein
Procarbazine (Matulane or Natulan)
Prednisone
Stanford V regimen
Doxorubicin
Vinblastine
Mechlorethamine
Vincristine
Bleomycin
Etoposide
Prednisone
Others:
Bulky disease - tumors at least 1/3 as wide as the chest or in other areas that are at least 10 cm across
Example of lymphocyte-depleted Hodgkin's, where background is rich in histiocytes, some small lymphocytes, and large RS cells

Immunodeficiency-associated lymphoproliferative disorders
Lymphoproliferative diseases (LPD) assoc c primary immune disorders (PID)
MC PID in LPD are ataxia telangiectasia (AT), Wiscott Aldrich syndrome (WAS) CVID, X-linked LPD (aka Duncan dz), Nimegen chromosomal breakdown syndrome
- LPDs in PID are DLBCL, LG, T-cell neoplasms
(same as Polymorphic B-LPD?) [23]
- Morphologically polymorphous lesions that efface overall tissue architecture and cause destructive masses, but do not fulfill the criteria for the diagnosis of lymphoma
- Most are genetically clonal proliferations drived by EBV that typically lack other genetic drivers
- driven by EBV in most cases
Polymorphic B-LPD in Autoimmune/Iatrogenic setting
- iatrogenic LPDs arise in patients treated with immunosuppressive regimens for autoimmune disease
- contribution of underlying autoimmune disease versus drug is difficult to separate, and may depend on disease severity
- the treatment regimens of a wide variety of solid tumors and hematologic malignancies can predispose to secondary IA-LPDs [23]
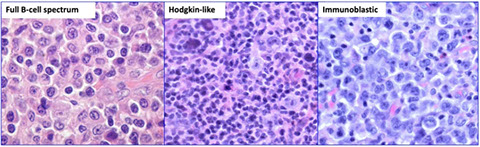

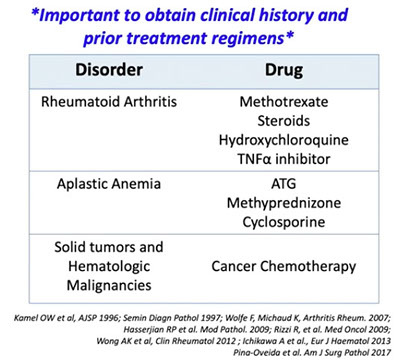
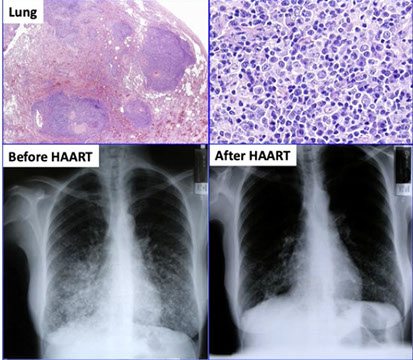
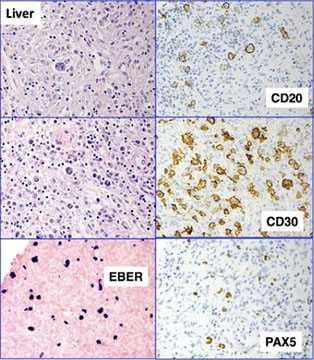


Polymorphic B-cell LPD. [23]
Polymorphic B-cell LPD features. [23]
Polymorphic B-cell LPD in the autoimmune setting. [23]
Polymorphic B LPD in HIV / AIDS patients. Lesions regressed after initiation of HAART therapy
Polymorphic B LPD in HIV / AIDS patients. Lesions regressed after initiation of HAART therapy
5 - 5
<
>
Lymphomas assoc c HIV infection
BL, DLBCL (esp in CNS), PEL, plasmablastic lymphoma, HL
Post-Transplant Lymphoproliferative disorders (PTLD)
Diseases ranging from mono-like sickness to neoplastic processes which can be due to EBV or not and occurs due to immunosuppression usually within 1 year after transplant
- lowest % in renal / BMT allografts, highest in heart/lung/ intestinal allografts, which in turn determines the amt of immunosuppression a pt will get;
- if pt EBV negative pre-transplant they are also at higher risk of dz; the PTLD clone usually of recipient origin but is seen in the transplanted organ itself
- commonly extranodal, esp occurring in GI, lungs and the liver
Incidence: ~3% of all transplant recipients but widely variable depending on risk factors
- lymphoma accounts for ~20% of cancers in solid organ transplant recipients, compared to 4-5% of cancers in immune competent adults [24]
- usually assoc c EBV, (60-70%)
- HHV-8 can occur later (after 5 years) and are more freq EBV-negative
-- can do serial EBV viral loads of serum
-- EBV-negative occurs later than those with EBV, and are less responsive to reduction in immunosuppression
__________________________________________
Risk factors:
- Age at time of transplant (children have higher incidence of PTLD than adults, more likely EBV naive)
- EBV status of donor and recipient
- type of transplant
- internsity of immunosuppression
- genetic polymorphisms leading in creased anti-inflammatory cytokines or decreased pro-inflammatory cytokines
- dono / recipient HLA-DR mismatch
Mechanism: impaired cytotoxic T-cell response leads to uncontrolled expansion of lymphocyets and plasma cells
- solid organ: recipient lymphocytes
- HSCT: Donor lymphocytes
__________________________________________
Clinical features:
- Majority within 1year of transplant
-- within 6 months of transplant for allo-HSCT
- second peak 5-15 years post-transplant
- EBV-negative cases: 4-5 years post-transplamnt
- T/NK cell cases: 6-7 years post-transplant
- Children have shorter interval from transplant to PTLD
- IM-like illness common
Sites of involvement: LN, GI, lung, liver
- often multifocal
- 20% of PTLDs after solid organ transplant confined to the allograft
Detection: EBV viral load: may increase in blood as disease develops and orogresses
- use of viral load for monitoring/diagnosis of PTLD is controversial
__________________________________________
Tx: restore anti-EBV T-cell immune response
- immunosuppresision reduction
Anti-neoplastic therapy:
- rituxumab
- cytotoxic therapy
- Trials: EBV-directed cytotoxic T-cells, mTOR inhibition, antivirals
__________________________________________
Px: pretty good

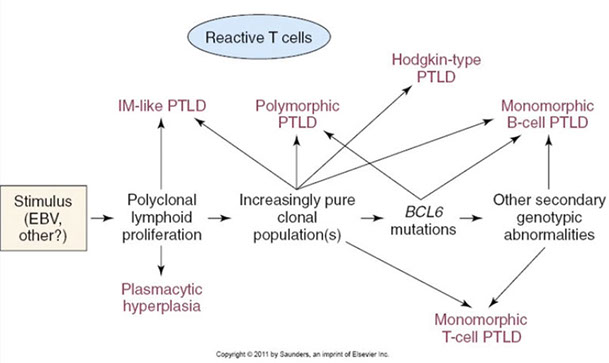
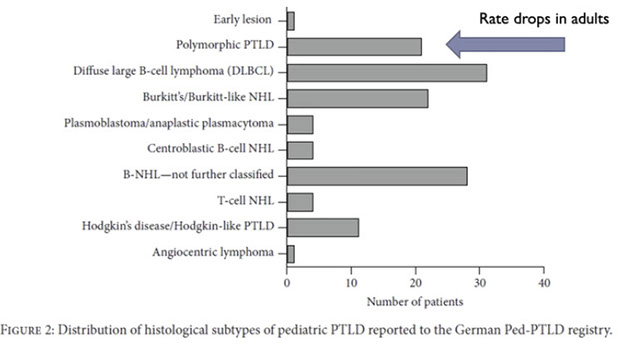


Risk of PTLD versus age, higher risk in the first year of transplant, kids 1-10 years of age with greatest risk, which goes back to the risk of their pears after 2 years of transplant. Babies <1 yo may still have protection from maternal antibodies [24]
The type of transplant dictates what kind of immunotherapy used, may partially account for these differences
May have lymphoproliferations that are or are not clo9nal.
PTLD types in pediatric population. Rate drop in adults
PTLD treatment
1 - 5
<
>
Polymorphic PTLD
- Architectural effacement / tissue destruction as would be seen in a lymphoma
- polymorphous cell infiltrate of immunoblasts, plasma cells, and variably-sized lymphocytes
Does not meet criteria for a specific type of lymphoma
Diagnosis:
- immunophenotyping shows a mix of T-cells and B-cells in varying proportions
- B-cells usually polyclonal but can see monoclonal populations
- largely a morphologic diagnosis (FISH, IHC, PCR, and flow will not help)
- should NOT do clonal gene rearrangement studies in cases of suspected polymorphic PTLD (may of may not be seen)
IHC: not really helpful, is more of a morphologic diagnosis, may be misleading
- CD30 may lead you to think that it is Hodgkin (and can even see a little CD15) but is NOT Hodgkin. Vs Hodgkins, you seem a spectrum of EBV+ cells, such as plasma cells and smaller cells

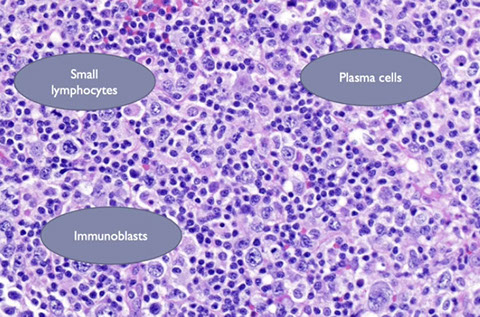

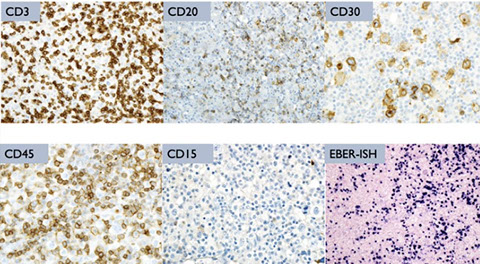
Polymorphic PTLD: key is architectural effacement
Polymorphic PTLD: has mix of cells
Polymorphic PTLD: has mix of cells, some are polymorphic and transformed
Polymorphic PTLD: has mix of cells, the CD30 may lead you to think that it is Hodgkin (and can even see a little CD15) but is NOT Hodgkin. Vs Hodgkins, you seem a spectrum of EBV+ cells, such as plasma cells and smaller cells
4 - 4
<
>
Non-destructive PTLD
- previously called "early" lesions
Includes:
- plasmacytic hyperplasia
- IM-like
- Florid follicular hyperplasia
Micro: Intact architecture, typically LNs or tonsils
- polytypic
Infectious Mononucleosis (IM)-like
- florid proliferation of small lymphocytes, plasma cells, and immunoblasts
- would be consistent with IM in a normal host
Plasmacytic hyperplasia
- proliferation of plasma cells and small lymphocytes
- have polyclonal IHC
Florid follicular hyperplasia
- - should make diagnosis cautiously because can be a normal reactive process in certain organs
- EBV-positivity can be found incidentally in reactive infiltrates of the GI or lung in peds
- no apparent impact on survival and no increased incidence of PTLD
- if considering a diagnosis of non-destructive PTLD with a florid follicular hyperplasia pattern, really make sure that there is a lot of EBV+ cells there [24]
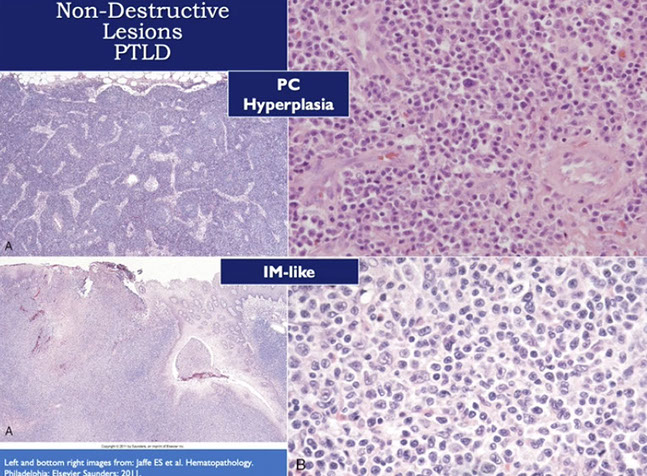
Monomorphic PTLD (- and T/NK- cell types) (M-PTLD)
- classified according to WHO 2016 nomenclature and disease definitions as for immunocompetent patients and must fulfill criteria for one of B-, T-, or NK-cell lymphoma
- nomenclature does not recognize the contribution of immunodeficiency
If you think to yourself that what you are looking at is a DLBCL in a post-transplant patient, it is probably monomorphic PTLD
Exception: Low-grade B-cell lymphomas are not considered PTLD even when they occur post-transplant. MALT lymphoma will now be included when EBV-posistive
- thought that these are just sporadically occurring and not associated with the transplabnt
Most are EBV+
Most have clonal gene rearrangement
Some have cytogenetic abnormalities (more common in early or polymorphic lesions)
Diagnosis as:
"Post-transplant lymphoproliferative disorder, monomorphic type, with features of ____ lymphoma"
Diagnostic workup should be same as in immunocompetent host
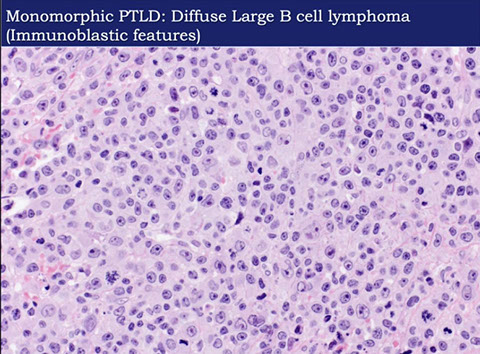

Monomorphic PTLD: DLBCL [24]
Monomorphic PTLD: plasmacytoma [24]
Monomorphic PTLD: Burkitt lymhpoma [24]
3 - 3
<
>
Monomorphic B-cell PTLD
-
Monomorphic T/NK-cell PTLD
-
Classical Hodgkin lymhpoma PTLD
- least common major form of PTLD
- Challenging bc HL-like cells occur in other types of M-PTLD and P-PTLD
- should be EBV+
- Should fufill strict criteria for CHL
- CD15 and CD30+, CD20-negative
- distinguish from Hodgkin-like PTLD
CHL-PTLD [24]

Other iatrogenic immunodeficiency-associated lymphoproliferative disorders
-
Histiocytic and dendritic cell neoplasms
Histiocytes
Includes [13]:
Macrophages: Phagocytosis; derived from bone marrow precursors and peripheral blood monocytes
Dendritic Cells:antigen presentation; derived from bone marrow precursors (usually); also circulate in peripheral blood (pretty few in number, may be able to catch them with flow cytometry)
Cyclin D1 is often aberrantly expressed in the nuclei of clonal / neoplastic histiocytic proliferations
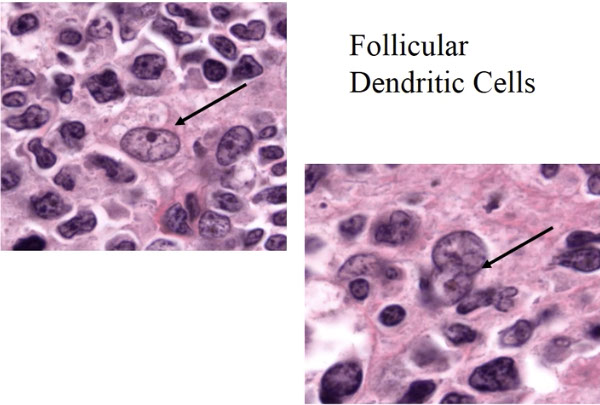
FDCs [13]
Finding interdigitating dendritic cells, look between 2 reactive nodules...
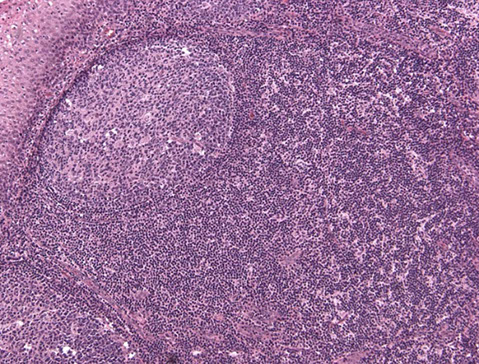
Normal interdigitating dendritic cells, look somewhat cleaved


BPDCN
Monocytic leukemias
Monocytic development and neoplasias
JXG
LCH
FDC
IDC
Histiocytic sarcoma
Histiocyte Society Classification [13] - WHO does not discuss reactive dz


Cyclin D1 is often aberrantly expressed in the nuclei of clonal / neoplastic histiocytic proliferations
Histiocytic sarcoma
- aka Kupffer cell sarcoma
Very rare cancer of cells that look like histiocytes
- gotta exclude acute monocytic cancers
- many entities previously thought to be malignancies of histiocytes are now known to be of other lineages
- generally presents in adults (median 61 years) with nodal or extranodal presentation
__________________________________________
Micro:
- poorly cohesive clusters of large cells with mild to marked nuclear atypia and abundant cytoplasm
- indistinguishable from DLBCL
- no Birbeck granules on EM
__________________________________________
IHC: (+) for 1 or more histiocytic markers such as CD163, CD68 or lysozyme
- may be positive for CD68, CD14, CD11c, lysozyme, CD45, HLA-DR, CD4, and is CD163+ in most cases
- negative for CD1a / langerin (Langerhans cells), CD30, CD21/35 (follicular dendritic cells), or CD33/13/MPO (myeloid markers)
__________________________________________
DDx: Poorly differentiated carcinomas, sarcomas
__________________________________________
Px: poor
- aggressive clinical course, often disseminated disease
Histiocytic sarcoma (Kupffer cell sarcoma)

Histiocytic sarcoma [13]

Tumors derived from Langerhans cells
Langerhans cell histiocytosis (LCH)
Lytic bone lesion made of of Langerhans cells in skull/vertebra/legs of white male kiddos that make CD1a, Cyclin D1, S100 and Langerin
- will see Birbeck granules on EM
aka Hand-Schuller-Christian dz (c multiple lesions) or Letterer-Siwe dz (c disseminated / visceral dz)
- Rare; ~5 per million per year (childhood); 1-2 per million per year (adult)
•Most common in 1-3 year olds.
•M:F 3.7:1
•More common in Northern European descent.
•May be localized, multiple within a system, or disseminated.
Associated with ALL, lymphoma
Identification of Langerhans cells in appropriate cellular background.
•Langerhans cells: oval nuclei with nuclear groove/fold and abundant cytoplasm; minimal or no cytologic atypia.
•Background: abundant eosinophils, admixed with neutrophils, and multinucleated giant cells; mild fibrosis.
•Lymph node: sinusoidal pattern.
•Other sites: nodular/patchy; variable extent of involvement.
- Langerhans cells can fill the papillary dermis and look like melanoma (like melanoma, Langerhans cells are also S100+)
Unifocal - most common form "Eosinophilic granuloma" -- in the bone, lymph node, skin, lung
Multifocal, multisystem disease - "Letterer-Siwe disease", bones, skin, liver, spleen, and lymph nodes, skull lesions, diabetes insipidus
Multifocal bone involvement - "Hand-Schuller-Christian disease"
__________________________________________
Pathogenesis
Derived from myeloid dendritic cells expressing CD1a and CD207 (not skin Langerhans cells).
•Clonal mutations involving genes in the MAPK pathway in >80% of cases.(??? may be more rare)
–BRAF V600E (~50%); can be detected by IHC
–MAP2K1 (~20%); WT BRAF
–ARAF, MAP3K1, ERBB3, PICK1, PICK3R2
•Other signaling pathways:
–PIK3CA, PICK1, PICK3R2
__________________________________________
LCH and "trans-differentiation" [13]
- association of LCH with lymphomas, esp follicular lymphoma (FL)
- cases shown to be clonally related (eg LCH harboring IGH/BCL2 fusion or IGH rearraangement by PCR)
- suggests that a differentiated lymphoma cell "trans-differentiates" into a Langerhans cell!!
- not clear if worse prognosis than de novo cases
__________________________________________
Micro: Langerhans cells have oval, groovy, folded nuclei with minimal atypia, variable mits, absent nucleoli, red cytoplasm and do not have dendritic cells processes (unlike epidermal Langerhans cells)
- background mix of eos, histiocytes, neuts, lymphs
- early on can see eosinophilic abscesses with central necrosis and Charcot-Leiden crystals
Birbeck granules on EM look like tennis rackets
__________________________________________
IHC: (+) CD1a (membranous c nuclear dot), S100, langerin (CD207), vimentin, CD68 and CD163, HLA-DR
- negative: CD45, lysozyme, CD21, CD35
__________________________________________
Genes: clonal by X-linked androgen receptor gene assay (HUMARA)
__________________________________________
Px: depends on staging, great for unifocal dz, worse if multifocal (involvement of multiple organs)
LCH lesions may precede or coexist with ~20% of cases of Erdheim-Chester disease.

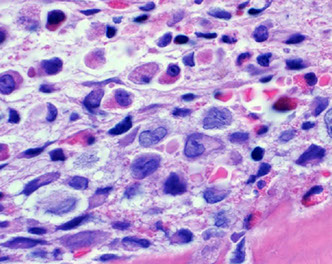
LCH

Birbeck granules in LCH


LCH - low power [13] - granulomatous-appearing infiltrate with lymphocytes and variably-sized histiocytes

S100 staining in LCH [13]

LCH molecular findings
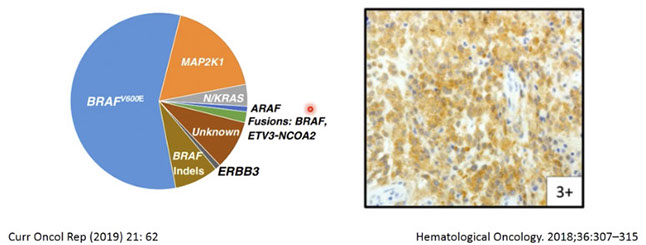
Langerhans cell sarcoma (LCS)
Rare high-grade cancer with crazy-lookin cells, usually in skin and multiorgans of adults c F>M
- some cells may look like Langerhans cells, on EM has Birbeck granules
IHC similar to LCH, S100+, but usually CD1a negative
Px: poor, 50% overall survival
[13] Case of histiocytic sarcoma arising from LCH, from EAHP workshop 2016

Indeterminate dendritic cell sarcoma
-
Interdigitating dendritic cell sarcoma
- Very rare
- Adults (mean age 55 yo)
Usually single lymph node enlarged, may be some extranodal cases
__________________________________________
Micro
Storiform proliferation or fascicles of spindled cells
- variable cytologic atypia
- look somewhat like follicular dendritic cell tumors
__________________________________________
IHC
Positive: S100, CD68, lysozyme, CD45
Negative: CD1a, CD21, CD35, MPO, CD34, B-cell or T-cell antigens, EMA, keratins
__________________________________________
Px
Benign in localized cases, lethal with widespread or disseminated cases
Interdigitating dendritic cell sarcoma [13] looks somewhat like FDC sarcoma, but positive for S100 and negative for FDC markers, with cleared out chromatin and small discrete nucleoli

Follicular dendritic cell sarcoma
- Rare neoplasm thought to be derived from follicular dendritic cells
Variable degrees of cytologic atypia and varied clinical course
Young to middle age (median 50 years)
- typically present with adenopathy, but many extranodal sites may be involved
- tonsil, spleen, oral cavity, others
May be associated with Castleman's disease in 10-20% of cases
_________________________________
Micro:
Sotriform proliferation and fascicles formed by plump to spindled cells
- usually bland cytology, but some may be atypical
_________________________________
IHC
May be positive for CD21, CD23, CD35, clusterin, vimentin, fascin, HLA-DR, EMA, CD68
Negative: CD1a, CD79a, CD30, HMB-45, keratins
Sometimes positive for CD45, CD20
- up to 50% have admixed T lymphoblasts ("indolent T lymphoblastic proliferation")
_________________________________
Tx/Px
Most cases indolent, may be treated by surgical excision
- local recurrence in 40-50%
Approximately 25% metastasize, with 10-20% dying of disease
A fairly typical case of follicular dendritic cell sarcoma, with eosinophilic cytoplasm and admixed lymphocytes, (right) nuclei resemble normal FDCs
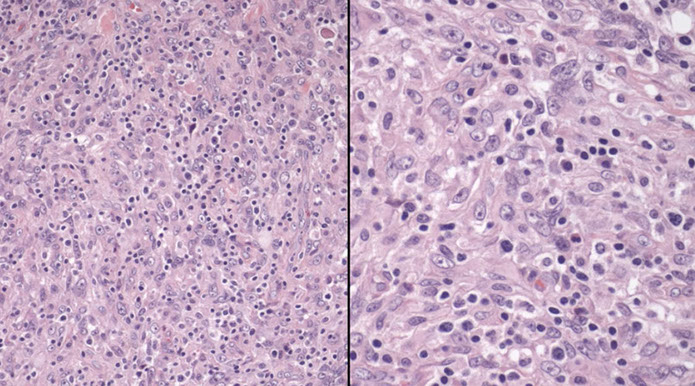
FDC sarcoma, CD21

Inflammatory pseudotumour-like follicular/fibroblastic dendritic cell sarcoma
-
Fibroblastic reticular cell tumor
-
Disseminated juvenile xanthogranuloma
-
Erdheim-Chester dz
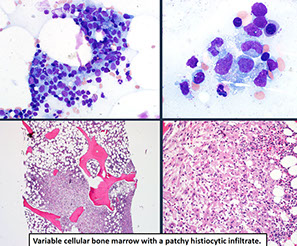
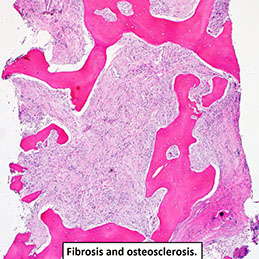

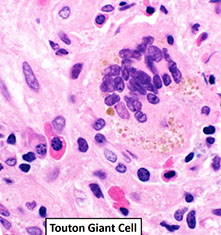

Erdheim-Chester disease
Newly added category of histiocytic/dendritic cell neoplasm in the revised 4th ed. (2017) WHO classification.
•Multifocal osteosclerotic lesions of the long bones (>95%)
•Median age 55-60 years; M:F 3:1
Frequently involves extraosseous sites: Heart (>50%), lung, maxillary sinus, large vessels, retroperitoneum, CNS, skin (30%; less common than LCH), pituitary gland, orbit.
•Heterogeneous clinical course.
•No standard treatment; 50% 3 year survival.
Extensive effacement by cytologically bland foamy histiocytes with interspersed inflammatory cells and multinucleated (Touton) giant cells.
•Fibrosis usually present and often prominent.
__________________________________________
Imaging:
Radiology highly suggestive of ECD:
- lower extremity long bones show symmetric osteosclerosis of diaphysis and metaphysis
__________________________________________
IHC
•Positive: CD68, CD163, FXIIIa.
•Negative: CD1a, CD207 (Langerin), S100-/+.
•Histopathologic features not specific for ECD.
•Definitive diagnosis requires correlation of pathologic features and clinical/radiographic findings.
granulomatous inflam, usually in long bones and spares the axial skeleton
__________________________________________
Genes:
Similar to LCH, all cases have at least 1 mutation leading to activation of the MAPK pathway.
•BRAF V600E mutation (~50%)
–Antibody useful in difficult diagnosis
–Treatment with BRAF inhibitors (vemurafenib) showed efficacy
•MAP2K1, NRAS, KRAS, ARAF, PIK3CA.
__________________________________________
Px
Chronic progressive disease
- outcome worse with more disseminated involvement
Some respond to interferon therapy
- BRAF, other targeted inhibitors may be effective
More recent survival data: 68% at 5 years
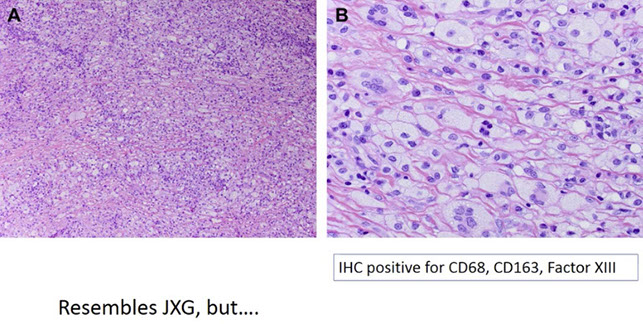

References:
1. Hutchinson, C et al. Orimary Mediastinal (Thymic) Large B-cell Lymphoma. Arch Pathol Lab Med. 2011; 135: 394-398
2. Johnson, Peter WM. Primary Mediastinal B-Cell Lymphoma. Hematology 2008
3. Joos, Stefan. Primary Mediastinal (Thymic) B-Cell Lymphoma is Characterized by Gains of Chromosomal Material Including 9p and Amplification of the REL gene
4. Dunleavy, K. Primary mediastinal B-cell lymphoma and mediastinal gray zone lymphoma: do they require a unique therapeutic approach?. Blood, 1 January 2015; vol 125 no 1
5. Sloan, C et al. Multifocal Mantle Cell Lymphoma In Situ in the Setting of a Composite Lymphoma. AJCP Feb 2015; p274-282
6. Hamadeh F et al. MYD88 L265 mutation analysis helps define nodal lymphoplasmacytic lymphoma. Modern Pathology April 2015. 28, 564-574
7. WHO Classification of tumors of hematopoietic and lymphoid tissues. 4th edition
8. Medeiros. Lymph Nodes and Extranodal Lymphomas. 2018.
9. Swerdlow S. Dealing wiht Small B-cell Neoplasms of Lymph Node and Pleen (Part 1). https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=416999412
10. Jaffe E. Hodgkin Lymphoma Classification and Differential Diagnosis. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=422552909
11. Sohani A. Aggressive B-cell lymphomas. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=427928433
12. Jaffe E. Peripheral T-cell and NK-cell neoplasms par1. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=445025939
13. Cook J. Introduction to Histiocytic disorders. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=414271529
14. Cook J. Plasma Cell Neoplasms. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=414271219
15. Amador. How I diagnose anaplastic large cell lymphoma. AJCP. 2021
16. Xie Y. How I Diagnose Angioimmunoblastic T-cell Lymphoma. Am J Clin Pathol July 2021:156:1-14
17. Lusky K. Lymphocytosis: distinguishing benign from malignant. CAP Today. May 2021.
18. Cherian S. How I diagnose minimal/measurable Residual disease in B lymphoblastic leukemia/lymphoma by flow cytometry. Am J Clin Pathol Jan 2021; 155:38-54
19. Gibson SE, Swerdlow S. How I diagnose Primary Cutaneous Marginal Zone Lymphoma. Am J Clin Pathol October 2020;154:428-449
20. YouTube. Approach to the lymph node. https://www.youtube.com/watch?v=EbZGWXY3m-s
21. Kelemen K. Eosinophilia / Hypereosinophilia in the Setting of Reactive and Idiopathic Causes, Well-defined Myeloid or Lymphoid Leukemias, or Germline Disorders. Am J Clin Pathol Feb 2021; 155:179-210
22. Crange G. PathPresenter Masterclass in Pathology. Approach to T-cell Lymphomas. https://conferences.pathpresenter.net/take-conference/9b5564f6ab83fe7bbb715df2481282866495c53d33c9f5484a9485714902f473CQByJg1UHTciiQ0Daq2Lnw==/read-lecture/550d8bc36c5b5ad4c0bc2e025799e04fa934c27d6726d74331abd0a58ff3e4daICGtv6HY%2FyvQtdX+uP+PJQ==
23. Natkunam Y. Immunodeficiency Associated Lymphoproliferative Disorders: Time for Reappraisal? Soc for Hemepath. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=412839740
24. King R. Post-Transplant Lymphoproliferative Disorders. Soc Hem Path.https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=421612096
25. ETP ALL Soc Hem Pathol Buddy Fuda. https://society-for-hematopathology.org/web/education-video-tutorials-view.php?video_id=348183825
26. Travaglino A et al. Prognostic Significance of CD30 in Transformed Mycosis Fungoides. A Systemic Review and Meta Analysis. Am J Clin Pathol 2021