
Liver
Gallbladder and Biliary System
Embryology, Anatomy, Histology
Liver Enzymes - see Enzmes
Normal biopsy
Patterns of Liver Injury
Hepatic Vascular Diseases
Iron Overload Disease
a1-antitrypsin deficiency
Hepatolenticular degeneration (Wilson's disease)
Diabetes Mellitus in Liver
Pregnancy and Liver Disease
Nutmeg liver
Hepatic steatosis
Alcoholic cirrhosis
Autoimmune Hepatitis
Acute and Chronic Viral Hepatitis
Hepatitis A
Hepatitis B
Hepatitis D
Hepatitis C
Hepatitis E
Other Viral Hepatidities
Adult Giant Cell Hepatitis
Spontaneous Bacterial Peritonitis
Other Infections of the Liver
Autoimmune Hepatitis
Drug Effects
Fatty Liver Disease
Granulomatous Disease
Liver failure
- Acute Liver Failure
- Chronic Liver Failure
Cirrhosis
Hepatic encephalopathy
Liver Transplant
Glycogen Storage Disease
Hepatic adenoma
Focal nodular hyperplasia
Macrodegenerative Nodule
Dysplastic Nodule
Hemangioma
Mesenchymal Hamartoma
Embryonal Sarcoma
Hepatoblastoma
Fibrolamellar hepatocellular carcinoma
Hepatocellular carcinoma
Clear Cell Carcinoma
Hepatic Progenitor Cell Carcinoma
Epithelioid Hemangioendothelioma
Embryology, Anatomy and Histology
Biliary Atresia
Neonatal Giant Cell Hepatitis
Paucity of Intrahepatic Bile Ducts
Familial Cholestasis
Cholecystitis
Gallstones
Hepatic abscess
Clonorchis sinensis and Opisthorchis viverrini
Reye's syndrome
Budd-Chiari syndrome
Veno-occlusive disease (sinusoidal obstruction syndrome)
Hemochromatosis
Acute Biliary Obstruction
Chronic Biliary Obstruction
Primary Biliary Cirrhosis
Secondary biliary cirrhosis
Primary Sclerosing Cholangitis
IgG4 Sclerosing Cholangitis
Caroli disease
Congenital Hepatic Fibrosis
Polycystic Liver Disease
Choledochal cysts
Physiologic neonatal jaundice
Gilbert's syndrome
Criggler-Najjar syndrome
Dubin-Johnson syndrome
Rotor syndrome
Banff Liver Rejection Schema
- acute rejection
Q fever
Bile duct adenoma
Biliary Adenofibroma
Simple Cysts
Bile Duct Hamartoma
Bile duct malformation (von Meyenburg complex)
Mucinous Cystic Neoplasm
Intraductal Papillary Biliary Neoplasm
Cholangiocarcinoma
Lymphoepithelioma-Like Cholangiocarcinoma (LELC)
Liver
Embryology
Appears in middle of 3rd wk as outgrowth of endodermal epithelium at distal end of foregut, called the hepatic diverticulum or liver bud, which penetrates the septum transversum (mesodermal plate bwt pericardial cavity and yolk sac)
- all of the foregut endothelium has potential to differentiate into liver cells, but is inhibited by surrounding tissues (esp the notochord), but these inhibitors are blocked by cardiac mesoderm production of fibroblast growth factor 2 (FGF2), Bone Morphogenic Proteins (BMPs) assist in this process
- bile duct forms as the tract of liver cells penetrating septum transversum narrows
- the gallbladder forms as a ventral outgrowth of the bile duct, and bile starts to enter the GI tract by the 12 WGA via bile duct
- epithelial cords mix c vitelline and umbilical veins and form hepatic sinusoids which differentiate into liver parenchyma and bile duct lining
-- hematopoietic cells, Kupffer cells and connective tissue cells develop from mesoderm of septum transversum
- the mesoderm of the septum transversum eventually becomes lesser omentum and falciform ligament (known collectively as the ventral mesentery)
- part of the septum transversum that is in contact with the liver becomes the central tendon of the diaphragm
- hematopoiesis declines in the last 2 weeks of life and is minimal at birth
Anatomy
Largest gland in the body (1400-1600 g), all nutrients absorbed in GI go through portal tracts to the liver
- the subphrenic recess (superior extensions of the peritoneal cavity found bwt diaphragm and diaphragmatic surface of liver, and is split into left and right sides by the falciform ligament, which projects anteriorly from the liver surface
- the hepatorenal recess (Morison pouch) is the superior-posterior portion of the subhepatic space, found bwt liver and right adrenal and right kidney
- diaphragmatic surface of the liver is covered c visceral peritoneum except the posterior "bare area of the liver" which is demarcated by the upward inflexion of the coronary ligament
- the right sagittal fissure and the umbilical (left sagittal) fissure are linked by the porta heptais on the inferior aspect of the lifer and form an "H"
-- the umbilical fissure formed anteriorly by the fissure for the round ligament and posteriorly by the fissure for the ligamentum venosum
--- the round ligament of the liver (ligamentum teres hepatis) is the fibrous remnant of the umbilical vein, which carried well-oxygenated blood from the placenta to the fetus
--- the ligamentum venosum is the fibrous remnant of the ligamentum venosum, the fibrous remnant of the fetal ductus venosus, which shunted blood from the umbilical vein to the IVC, short-circuiting the liver
- the lesser omentum encloses the portal triad (bile duct, hepatic artery and hepatic portal vein) and goes from the posterior surface of the liver to the lesser curvature of the stomach and the first part of the duodenum
- functionally the liver is divided into left and right by the branching of the portal triad into the left and right branches, except for the caudate lobe (segment I), the separating line is sometimes known as the Cantlie line, although the left medial division is considered part of the right anatomic lobe
- liver divided by the fissures, peritoneal reflexions and vessels into 2 anatomic lobes and 2 accessory lobes
-- falciform ligament separates the liver into left and right lobes
-- accesory lobes are the anterior-inferior quadrate lobe and the posterior-superior caudate lobe
Blood supply to the liver is dual, coming from the hepatic portal vein (which has ~40% more oxygen than systemic blood returning to the heart), and the hepatic artery (from the celiac trunk), which only supplies ~1/4 of the liver's needs
- portal vein and hepatic artery enter liver via inferior part of liver through hilum, the porta hepatist
- the hepatic portal vein is formed by the superior mesenteric and splenic veins posterior to the neck of the pancreas
- most of the lymph drains as superficial lymphatics in Glisson capsule
-- lymph forms in the perisinusoidal spaces of Disse (beneath the endothelial cells) and drain to the deep lymphatics in the intralobular portal triads
- nerves to the liver come from the hepatic plexus, the largest derivative of the celiac plexus
Histology
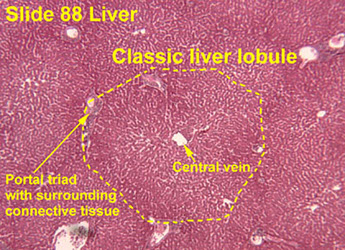
Broken into hepatic lobule (2D) or acinus (3D), where the hepatic lobule has central venule with hepatocyte cords radiating to peripheral portal tracts
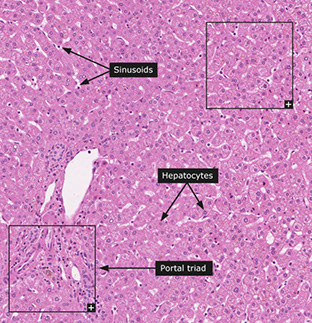
Portal Tracts
Contain hepatic artery, portal vein, and bile duct
- hepatic artery is normally about the same diameter as the bile duct, and are seen together most of the time, while the portal veins have ~5x the lumen diameter
- 1/10 of the smallest branches of the portal tracts do not have a bile duct on H&E normally
-- medium and larger portal tract should always have a bile duct
Lobules
Made of cords (plates) of hepatocytes usually 2-3 cells thick
- hepatic plate thickness best seen c reticulin stain
- hepatocytes have blood flow on both sides, one from portal venous blood and other from blood from hepatic artery
- sinusoids lined by fenestrated endothelial cells
- under endothelial cells is the space of Disse, that extend into hepatocyte microvilli (space of Disse also contains fat-containing myofibroblastic hepatic stellate cells)
- Kupffer cells are scattered on the luminal surface of endothelial cells
- between hepatocytes are bile canaliculi (separated from vascular space by tight junction) that drains into canal of Hering, which then goes to bile ductules in periportal region, that empty to terminal bile ducts in the portal tracts
Lobes divided into zones based on proximity to normal structures
- Zone 1: hepatocytes around portal tracts (these cells have more rich O2 and nutrient supply than zone 3)
- Zone 2: cells not in zone 1 or 3
- Zone 3: hepatocytes around the Central vein (C=3) and are important in detoxification
Central vein
vary in size (larger at hilum, smaller at periphery of liver), which corresponds to the thickness of the wall


CV




Normal biopsy
Most places do a couple H&Es, trichrome and iron, and some also include PAS and reticulin on all medical liver biopsies
- hx is important (understand why the bx performed)
- adequacy determined by what you are looking for and what you see, but may be determined by length if involved in research, though a general guideline is to have >10 portal tracts and be >1 cm length
- sampling error is a function of size relative to the heterogeneity of the bx (ie more heterogeneous processes less likely to be adequately sampled)
- patterns of injury include: lobular hepatitis, portal-based chronic inflam, fatty change, bile tract obstruction / inflam / injury, bland lobular cholestasis, necrosis, abnormal inclusions / pigment, granuloma, vascular dz, infiltrates, and tumors
-- should recognize the most prominent pattern to aid in dx
-- being systematic (ie starting at portal tracts and working towards central vein) helps avoid errors
Bile duct proliferation usually caused by downstream bile duct obstruction (such as by stones, tumor, etc) or in acute viral hepatitis
Fibrosis from chronic injury ranges from no fibrosis, portal fibrosis, bridging fibrosis, and cirrhosis
Important to address in report the reason why the liver tissue was taken, and also the amount of fibrosis and inflam
- amt of fibrosis important for px in chronic HCV infx
Sinusoidal changes
Resolving Hepatitis Pattern - Lobular macrophages
Minimal to no inflam, with scattered small clusters of macrophages in the lobules (macrophages trying to eat away previous liver damage)
- can be seen in drug toxicity, viral-hepatitis
Hyperviscosity syndromes
Not well described, but can be seen in several hematologic malignancies, or autoimmune disorders
- can see subtle inflam, possible sinusoidal dilatation and congestion, or lobular distortion and nuclear pleomorphism (possibly 2/2 ischemia)
Sinusoidal dilatation
Can be 2/2 outflow obstruction; dilation usually in zone 3, and may have Kupffer cells c inc iron deposition and zone 3 central vein fibrosis if chronic
- if sinusoidal dilatation is present and no outflow dz or pertinent hx, may consider, may consider autoimmune dz, infx granulomatous dz, paraneoplastic dz, systemic inflam dz, or possibly from rapid blood vol expansion
- higher chance that sinusoidal dilatation is real if accompanied by hepatocyte atrophy
Hepatocyte changes
Prominent Megamitochondria
Assoc c fatty liver dz and cholestatic liver dz
- overall is a non-specific finding that can be assoc c drug rxn in adults or metabolic disorders in kiddos
Pseudoground Glass Change in Cytoplasm
Can be easy to spot in most cases, or can be more subtle
- usually have well-demarcated inclusions
Induced Endoplasmic Reticulin Proliferation
Hepatocytes have distinct amphophilic cytoplasm
- is assoc c various meds
Minimal Bland Lobular Cholestasis
MC c med rxns, but the differential is long
- copper stain can be very focal and mild, so watch out!
- CK7 can show mild staining in zone 1 in chronic cholestasis (is normally negative)
- suspect heart dz if CK7 positivity is in zone 3
Cryptogenic Fibrosis / Cirrhosis
If only pattern seen is fibrosis / advanced cirrhosis in a bx taken for unexplained chronic liver enzyme elevation
- differential includes a1-AT def, bile duct loss, Wilson dz (in younger pts), hep B/C. fatty liver dz, autoimmune dz, and several others
- telomere shortening syndrome causes cryptogenic fibrosis and unexplained pulmonary fibrosis
Patterns of Liver Injury
Age-related changes
Liver vol dec up to 40%, 2/2 dec in hepatocyte number and size
- blow flow also dec to liver accordingly
- hepatocytes get inc lipofuscin, and less smooth endoplasmic reticulum
- hepatocytes lose the ability to proliferate somewhat
Biliary Obstructive Pattern
2/2 obstruction of large branches of the bile duct
- pt usually presents c biliary colic (episodic abd pain)
- see bile ductular prolif, mixed inflam, and edema in portal tracts
-- can also have mild inc lymphs and apoptotic bodies in the bile duct epithelium
- may see ductular, canalicular or lobular cholestasis
Chronic biliary obstructive dz MC in PSC, chronic panc dz c duct strictures, or anastamotic strictures after transplant
- mild chronic portal inflam commonly seen
- obliteration of the ducts c fibrous tissue possibly in chronic extrabiliary strictures
Bland Lobular Cholestasis
Has little or no portal tract changes, very mild inflam, no inc fatty change
- seen mostly in hepatocytes or bile canaliculi
- MCC is drug effect if presenting as an acute hepatitis
-- also seen in septic pts
Fatty Liver
Macrovesicular steatosis MC (2/2 both alcoholic and non-alcoholic FLD)
- also 2/2 metabolic dz and malnutrition
Microvesicular steatosis shows hepatocytes filled c multiple little fat droplets
- is a result of mitochondrial toxicity, usually from a drug effect such as alcoholic foamy liver degeneration or fatty liver of preg
- should consider congenital metabolic effects in kiddos
- can also been as a reactive pattern after massive liver necrosis
Hepatitic Pattern
Very common. shows lobular inflam of mostly T cells, which are mild to markedly inc, can have scattered apoptotic or ballon hepatocytes, and confluent necrosis in zone 3 if severe
-- confluent necrosis occurs c widespread parenchymal loss and severe zonal loss of hepatocytes, resulting in a space full of cell debris, macrophages, and a reticulin meshwork remnant
-- bridging necrosis the zone of necrosis links central veins to portal tracts
- may show lobular disarray, where hepatocytes do not nicely line up in rows or cords
- portal tracts show inc lymphs (B and T) mixed c other chronic inflam and eos
- can have ductular proliferation in marked acute hepatitis, mimicking downstream biliary tract dz
Evaluating the Differential
Plasma cells suggestive of autoimmune dz (can also be seen in acute viral hepatitis (A or B)
- eos suggest allergies or medication rxn (tho most med rxns usually just have inc lymphs)
- Hepatitis E shows cholestatic hepatitis c neuts in the lobules
- EBV has sinusoids densely packed c lymphs, sometimes lined up in "beaded" pattern
- zone 3 hepatitis c mild lymphocytic venulitis seen in autoimmune dz, drugs and acute viral hepatitis
Giant Cell Transformation Pattern
Nonspeciic reactive change in hepatocytes
- can dx neonatal giant cell hepatitis or adult giant cell hepatitis
-- mild giant cell hepatitis can be seen in several conditions (cholestasis, drugs/herbs, hematologic malig, necrobiotic xanthogranuloma, genetic causes, autoimmune, infx)
- may be made by a fusion of hepatocytes, and are not proliferative
Bland Necrosis
Assoc c acetaminophen tox or ischemia
- can be panacinar or limited to zone 3
- in hepatocyte necrosis, cell swells 2/2 defective osmotic regulation at cell membrane, rupturing the cell
-- before rupture forms membrane blebs that carry off cytoplasm and cause macrophage response
- can have inc iron in Kupffer cells, or in ductules if proliferating
- cytoplasm of dead cells is more red, and the color change caused by necrosis usually most prominent around zone 3 (though appears more diffuse in cirrhotic pts)
- zone 1 necrosis rare, but assoc c halothane (tho more commonly causes zone 3 necrosis), proteus vulgaris endotoxin release, allyl alcohol
Hepatocyte apoptosis causes cell to shring, pyknosis (nuclear chromatin condensation), karyorrhexis (nuclear fragmentation) and formation of apoptotic bodies (aka Councilman bodies or acidophil bodies)
Vascular injury pattern
1. Obstructive pattern - seen in Budd-Chiari sundrome, chronic heart dz
2. Inury to endothelium of veins, causing thrombosis and fibrosis
- can be 2/2 chemo, clotting disorders, infx, EtOH, inflam central vein injury, viruses, drugs
3. Vascular-flow related path causes loss of portal veins
- may cause generalized liver atrophy
Chronic liver inury patterns
many cases of chronic liver injury lack fibrosis, several other patterns can be seen
Ductopenia
Loss of intrahepatic bile ducts, aka vanishing bile duct syndrome
- seen in chronic biliary tract dz such as PSC or PBC, chronic rejection of liver allograft, drug rx, paraneoplastic syndromes
Fibro-obliteratic Duct Lesions
Round / oval fibrous scar that has replaced a bile duct
- seen in chronic obstruction of an extrahepatic bile duct
- onion skinning seen early in the dz, c bile duct surrounded by dense collar of lamellar fibrosis (be careful bc normal large-sized bile ducts can look similar, though the look more atrophic in true onion skinning)
Iron or copper build up in hepatocytes
Copper assoc c Wilson dz, or chronic cholestasis
- copper staining in cirrhotic livers is less specific for chronic biliary tract dz as primary process
- usually seen c rhodanine stain
- copper can build up in PBC, PSC, other chronic cholestatic conditions
- also seen in focal nodular hyperplasia from cholestasis
- neonatal liver can also normally be pos for copper
Fibrosis Evaluation
Adequacy
Small bx c definite cirrhosis is adequate
- the bigger the better, though as a general rule should have at least 10 portal tracts and be 1 cm in length
Basic Fibrosis Patterns
can be seen in the portal tracts or hepatic lobules
- Lobular fibrosis MC in fatty liver disease, drugs, or chronic congestive liver dz
- Portal-based fibrosis can be seen in any chronic liver dz, beginning with portal fibrosis (as an expansion of the portal tracts)
Hepatic stellate cell is principal cell that forms scar deposits
- the stellate cell is a vit A (lipid) storage cell, but in severe acute and chronic injury the cell becomes active and are highly fibrogenic myofibroblasts
- PDGFR-B expression in stellate cells is inc
-- Kupffer cells and lymph excrete substances that regulate gene that control fibrosis in stellate cells
Fibrous septa form from areas of parenchymal loss where stellate cells activated in areas where reticulin layer is collapsed and hepatocytes have disappeared
- fibrous layer encircles surviving hepatocytes at later stages of injury
- scar layers become condensed and thin if injurious mech to liver has ceased, and can be broken down by hepatocyte metalloproteinases thus reversing fibrosis
Diagnosing fibrosis
Portal fibrosis diagnosed by assessing relative size of portal area and the smoothness of the border bwt portal tract and lobules
- can have:
1) global expansion of portal tract,
2) an irregular border of a fibrotic portal tract c fibrous extensions into lobules,
3) hepatocytes entrapped by fibrosis
Bridging fibrosis
Abnormal fibrous tissue between portal tracts, or from portal tract to central vein
- should be a little bit thick, thin broken strands may not count
Cirrhosis
Regenerative nodules of hepatocytes surrounded by fibrotic bands
- if nodularity is not complete, may diagnose early cirrhosis
Intralobular fibrosis called pericellular / perisinusoidal fibrosis
- can be seen in many diseases
- can call mild if only seen on trichrome, and moderate if seen on H&E
Fibrosis Staging Pitfalls
- Inflammation or bile duct prolif can mimic portal fibrosis
- bridging necrosis can mimic bridging fibrosis, and fibrosis staging in the setting of extensive necrosis can be inaccurate
- fragmented biopsies commonly seen in setting of advanced fibrosis and can also underestimate the degree of fibrosis
- "fibrous caps" seen around some nodules, and is indicative of bridging fibrosis, and sometimes cirrhosis
- portal tract branch points can mimic fibrous bridges
Fibrosis Regression Pattern
Fibrosis and cirrhosis can go away if the offending toxin is removed
- delicate fibrous bridges
- thick isolated collagen bundles in lobules
- spikes of fibrosis coming from portal tracts
- remnants of portal tracts or central veins
- minute regenerative nodules
- med-sized portal tracts and central veins in close proximity
Hepatic Vascular Diseases
Congenital / Genetic Abnormalities
Most share common elements: absent/atrophic portal veins, nodular regenerative hyperplasia, abnormal arterialization of portal tracts and lobules, patchy bile ductular prolif that looks like downstream biliary tract dz, developments of focal nodular hyperplasia
Abernethy Syndrome
Absent portal vein from birth c shunting of intestinal and splenic blood around liver to renal or hepatic veins
- micro: absent portal veins in small and medium-sized portal tracts c occasional hypoplastic portal veins in larger portal tracts
- nodular regenerative hyperplasia possibly seen
- hepatic arteries usually hypertrophied c prominent muscular coats
- mass esions can develop, such as focal nodular hyperplasia and HCC in non-cirrhotic livers
VATER Syndrome
Vertebral anomalies, Anal atresia, Tracheal-Esophageal fistula, Renal and Radial defects (VATER), also known as Vertebral anomalies, Anal atresia, Cardiac defects, Tracheal-Esophageal fistula, Esophageal atresia, Renal and Radial defects, Limb defects (VACTERL) syndrome
- caused by undefined defects in development of embryonic mesoderm
- unknown etiology, probably multifactorial
- liver can show absent/atrophic portal veins c inc prominent arterioles in portal tracts
- liver findings similar to Abernethy syndrome (focal nodular hyperplasia)
Turner Syndrome
Many have abnormal liver enzymes, and bx can show steatosis / steatohepatitis, and abnormal vasculature, with portal veins absent or atrophic
- may see nodular regenerative hyperplasia and focal hyperplasia
- bile ductular prolif can mimic biliary obstruction, and some pts can get cirrhosis
Hereditary Hemorrhagic Telangiectasia (HHT)
Liver can show AVMs in up to 3/4, telangiectasia in 1/2
- large AVMs can cause high output cardiac failure from blood shunting
-- bx usually not done 2/2 risk of bleeding
Hepatic Inflow Abnormalities
Portal Vein Thrombosis and Hepatoportal Sclerosis
Can be clinically occult and cause portal HTN and ascites
- many possible causes, 1/2 remain idiopathic, but pt should be worked-up for thrombotic disorders
- other causes include inflammatory conditions such as pancreatitis, appendicitis, and diverticulitis, prior abdominal surgery can predispose
- Micro: peripheral liver bx can have subtle changes, with the overall picture referred to as hepatoportal sclerosis (has atrophic or absent portal veins, herniation of portal veins to zone 1 hepatocytes, dense fibrous scars replacing portal veins, liver parenchyma c nodular regenerative hyperplasia, recannulated thrombi)
- portal biliopathy - assoc of portal vein thrombi c strictures and irregularities of extrahepatic bile ducts; is seen in non-cirrhotic livers usually and assoc c thromboses extension to mesenteric veins and thought to develop as collaterals to thrombosed veins; dx usually made on imaging, bx is rare
Hepatic Artery Thrombosis
Usually caused by thrombi, but can be related to vasculitis or anastomotic strictures
- blood from collateral portal circulation can compensate for thrombi in extrahepatic arteries
Sinusoidal Disease
- sinusoids are physically blocks, impairing blood flow
Sinusoidal Obstructive Syndrome (SOS)
- previousy veno-occlusive disease (bc veins not always occluded)
- 2/2 toxic or inflam injury to endothelium of sinusoids or central veins
-- up to 3/4 have obliteration of central vein
- possibly 2/2 herbal teas/remedies, bone marrow transplant, chemotx (oxaliplatin for colon ca), radiation
- can be overinterpreted by pathologists
- Micro: sinusoidal dilation and congestion, usually in zone 3 c "bridging congestion" on larger bx / resection
- can show inc iron in Kupfer cells in long-standing dz
- mild or no inflam in lobules, central veins can show fibrous obliteration, and can see nodular regenerative hyperplasia and peliosis hepatis
- cautery can cause dilation artifact in wedge bx, which have gray material in lumen
Sickle Cell Disease
- MC pattern is iron overload 2/2 transfusion, in Kupffer cells, can cause cirrhosis in up to 1/5 pts
- bx can give info on amt of fibrosis; can also see congested sinusoids, and erythrophagocytosis
- sickle cell crisis can cause cholestasis, sinusoidal congestion, and ischemic necrosis
Hemophagocytic Syndrome
- caused by severe hyperinflammation c uncontrolled prolif of macrophages, clinically causing fever, cytopenia(s), and HSmegaly
- get inc ferritin, alk phos, triglycerides
- GLUT1 stain can highlight RBCs in macrophage cytoplasm
- sinusoids are at least mildly congested
Vascular Outflow Disease
- core patterns is sinusoidal dilation, sinusoidal congestion, hepatocyte atrophy or dropout in zone 3, and zone 3 fibrosis in long-standing cases
- can be mild, patchy bile ductular prolif in portal tracts c mild mixed inflam, and can have overlap c SOS on micro (differentiate c hx, imaging)
- patchy mild sinusoidal dilation can be 2/2 inc fluids at time of bx
Budd-Chiari Syndrome
- hepatic veins occluded, with the exception of heart dz and sinusoidal obstructive syndrome (SOS)
- sx are abd pain, enlarged liver, and ascites
- MCC is thrombosis of hepatic veins 2/2 myeloproliferative disorder or clotting disorder, and can be caused by compression by a neoplasm
- Micro: thrombi not typically seen on peripheral liver bx, but instead see sinusoidal dilation and congestion
- zone 3 hepatocytes usually atrophic, and can have hepatocyte dropout
- acute obstruction can cause extravasation of RBCs to space of Disse
Heart Failure
Congestive heart failure or chronic lung dz c right-sided heart failure can cause congestion, with dilated and congested sinusoids and atrophic zone 3 hepatocytes
- zone 3 has hepatocyte dropout and fibrosis if long-standing
- commonly see Kupffer cell hyperplasia and iron deposits
- pseudo-ground glass inclusions can be seen
Other Cuases of Sinusoidal Dilation
Drugs (estrogen, chemotx, azathioprine), Autoimmune dz (RA, APS, Takayasu, Castleman, Crohn, Still dz), Paraneoplastic syndromes (RCC, Hodgkin), Systemic infx (HIV, brucellosis, TB), Others (heroin use, sickle cell anemia, hemophagocytosis)
Architectural Changes and Tumors Associated with Vascular Flow Abnormalities
Nodular Regenerative Hyperplasia
Develops in livers c altered blood flow patterns, c changes caused by ischemic atrophy and secondary nodular hyperplasia
- assoc c inc alk phos but normal AST/ALT
- has lots of assoc conditions (portal HTN c varices, ascites, Smegaly)
- called nodular transformation when nodularity is heavily accentuated
- can mimic cirrhosis grossly and on imaging, though there should not be much if any fibrosis
Micro: hepatic parenchyma with diffuse nodularity from small atrophic hepatocytes in zone 3, with normal to slightly big zone 1 and 2 hepatocytes, which is highlighted c reticulin stain
- may have portal vascular changes (hepatoportal sclerosis or portal vein wall hypertrophy)
- may develop after liver transplant, reason unknown, maybe azathioprine
• Hepatocellular nodules without fibrosis
– Hepatocytes between nodules are small, flattened, 2 cells thick
– Reticulin network compressed between nodules
IHC: No fibrosis on Masson trichrome
Peliosis Hepatis
- localized areas of sinusoidal dilation of variably-sized cavities, causing blood-filled pools / lakes
- liver usually enlarged and diffusely involved, and can affect spleen
- MCC are chronic debilitating dz's (TB, cancer), drugs, bacillary peliosis assoc c Bartonella henselae
- vascular pools can have liquid blood, though can see early thrombosis and organization at edges
- no cirrhosis usually seen in background liver
Macroregenerative Nodules
- can be seen c chronic vascular outflow dz (Budd-Chiari) or other causes of vascular cirrhosis
- can be seen in livers after massive necrosis
Hepatic Adenomas
- develop in chronic vascular dz (Budd-Chiari syndrome), usually c OCP use
Iron Overload Disease
Normal Iron (Fe) Metabolism
- Fe toxic if too much, but need it still
- 3-5 g in normal pt (a nickel if 5g); need 20 mg/day to function, majority of this met by iron release from normal RBC metabolism, rest is absorbed from sm intestine (where iron levels are ultimately regulated)
Hemosiderin: abnormal iron deposits
Proteins:
Divalent Metal Transporter (DMT)-1: transporter for iron from lumen of sm intestine to cytoplasm of enterocytes
Ferritin: major storage form of iron; huge iron storing capacity
Ferroportin: transportation of iron from enterocyte, macrophage, and hepatocytes to blood
Hemojuvelin: interacts c BMP and SMAD pathways c the downstream target hepcidin
Transferrin: transports iron in blood
HFE: protein similar to MHC-1 which interacts c transferrin receptor 1 to regulate hepcidin levels; mutations cause hemochromatosis
Cells:
Enterocyte: involved in short term iron storage, and also in Fe absorption
Hepatocyte: produces ferritin and hepcidin; stores iron as ferritin (major source of Fe)
Macrophage: recycles old RBCs; also stores some Fe as ferritin
Fe Absorption
Takes place in duodenum and prox jejunum
- Fe from heme comes from dissociation of heme from globin which is then eaten up by enterocytes
- non-heme iron must be reduced to ferrous Fe before it can be taken up by enterocyte cytoplasm by DMT-1
- after entering entocytes, can be transported out by ferroportin with the help of ceruloplasmin and hephaestin
- is bound by transferrin in blood
- if iron stores are sufficient, iron stored in enterocyte cytoplasm
- hepcidin degrades ferroportin, blocking Fe transport into blood
- when enterocyte dies, iron secreted into feces, lowering Fe levels
Healthy pt have more transferrin than Fe, and ~1/3 of transferrin saturated; transferrin can absorb excess Fe if levels rise
Fe Storage
Fe stored as ferritin in hepatocytes and macrophages
- ferritin can store ~4.5 k Fe atoms, though is usually not visible on iron stains, but can sometimes be seen as a blush in hepatocyte cytoplasm
- hemosiderin deposits can develop if ferritin elevated chronically, which are gold-brown deposits on H&E
- become anemic if iron stores insufficient, whereas Fe overload can develop if Fe metabolism outta whack the other way
Fe Release from Cellular (Enterocyte, Liver, Macrophage) Storage
Hepcidin major regulator of Fe metabolism, blocking Fe release
- more Fe gets absorbed from gut enterocytes if needed
- hepcidin coded by (HAMP gene), and also functions as an acute phase reactant when released by hepatocytes and bile epithelium (stops Fe release so bugs don't get it?)
- hepcidin stops Fe release by degrading ferroportin (the only known Fe export protein)
Detecting Fe in Liver
Iron Stain
Prussian blue stain turns ferric iron to ferric ferrocyanide, an insoluble blue compound that looks blue
- not sensitive to detect low iron levels
- can use scoring system from 0-4 to score iron levels
- hepatic iron index traditionally used to interpret quantitative Fe levels
Micro: iron usually in zone 1 hepatocytes first, c gradient from zones 1 to 3, and usually clusters around canaliculi ( a pattern seen in other non-HFE mutated condition), likely 2/2 hepcidin dysregulation
- iron can be in bile ducts or in endothelium, can be a common finding
Ferritin: normally cannot see ferritin, but if inc may see bluish blush
Hemosiderin: brown granular cytoplasmic deposits on H&E
- bright blue granular staining
Inc Fe in an explanted liver may be sign of dec survival for recipient
- iron deposits seen in ~1/3 of liver bx's from pts c chronic HCV, may be assoc c inc fibrosis stage
- up to 2/5 liver bx for NAFLD have Fe deposits, similar situation as chronic HCV
- EtOH inhibits hepcidin, this chronic alcohol abuse assoc c inc Fe
- marked Fe accumulation assoc c inc risk HCC
Fe-related Gene Mutations
Hepcidin essential in all kinds of hemochromatosis, which all have dec or impaired hepcidin function
- HFE, HAMP, HJV and TFR2 genes may all be affected
- dec hepcidin levels causes Fe depositions in liver and other organs
- if (rare) mutations cause inc hepcidin lvels, may get congenital refractory anemia, and may be the cause of hepatic adenomas
-- adenomas in liver in pts c type 1a glycogen storage dz may resolve a chronic anemia if adenoma excised
- HCC may dec hepcidin levels
Genetic Iron Diseases
Hemochromatosis type 1
AR, HFE gene, late onset
- C282Y (up to 9/10) and H63D (~1/20) are the MC mutations
-- C282Y assoc c European ancestry
- sx variable and may present c vague fatigue and bone pain, classic triad of bronze diabetes and cirrhosis not so common now 2/2 early dx
- HFE mutations also assoc c more severe morbidity if assoc c another chronic liver dz
Iron: Hepatocytes > Kupffer cells
Hemochromatosis type 3
AR, TFR2, early onset
- rare in Western population, variable clinical course
- TFR2 gene mutations commonly seen in asyx adults, sometimes c only modest Fe inc
Iron: Hepatocytes > Kupffer cells
Juvenile Hemochromatosis type 2A
AR, HJV (hemojuvelin gene), early onset
- rare, but is MCC juvenlle hemochromatosis; MC mutation is G320V (up to 9/10)
- presents as impotence or amenorrhea or cardiomyopathy
- Micro: hepatocytes can show very inc iron stores
- Px: severe course, can progress rapidly
Iron: Hepatocytes > Kupffer cells
Juvenile Hemochromatosis type 2B
AR, HAMP (hepcidin gene), early onset
- marked hepatocellular overload, severe course
- assoc c hypogonadism, cardiac dz
Iron: Hepatocytes > Kupffer cells
DMT-1 hemochromatosis
AR, SCL11A2 (DMT-1 gene), early onset
- very rare, data limited, may present c microcytic anemia
Iron: Hepatocytes > Kupffer cells
Ferroportin disease type B
AD, SLC40A1 (ferroportin gene), late onset
Iron: Hepatocytes > Kupffer cells
Diseases c Fe Deposits mostly in mesenchymal cells
- ferroportin dz is the classic example, Fe mostly in Kupffer cells
- inc transferrin levels not seen until later in dz
- type A dz has Fe in Kupffer cells, type B in hepatocytes
Ferroportin disease type A
AD, SLC40A1 (ferroportin gene), late onset
- may have small sideronecrotic foci c Fe-laden macrophages in small clumps in lobules
Iron: Kupffer cells > Hepatocytes
Hypotransferrinemia
AR, Tf (transferrin gene), early onset
Iron: Kupffer cells > Hepatocytes
Hypoceruloplasminemia
AR, CP (ceruloplasmin gene), late onset
Iron: Kupffer cells > Hepatocytes
Neonatal Hemochromatosis
- broadly classified as liver dz in neonate c inc Fe deposits
- most 2/2 alloimmune gestational dz c maternal abs crossing placenta and attacking fetal liver in utero (mech not very clear)
- neonate often c liver failure at birth or first days of life, may cause in utero fetal loss
- extrahepatic Fe deposits helpful in making dx, esp in pancreas, myocardium, thyroid, minor salivary glands, Brunner glands, pancreatic islets, stomach and chondrocytes
- BM and spleen often normal in terms of Fe
a1-antitrypsin (AAT) deficiency
AR inheritance of codominant trait; mutated a1-ATZ allele cr 14; normal allele designated "M"; while "Z" is the MC point mutation that leads to AAT in SERPINA1 at Glu342Lys Protease Inhibitor (Pi) locus
- "S" mutation not assoc c inc risk of emphysema, but does have slightly lower levels of AAT
- "M" comes from migration of a sample to the "M"iddle of an electrophoresis gel, where A-L is proximal and N-Z is distal to the M; thus "Z" would migrate farthest from the "M"iddle, and "S" not as much
- AAT normally inhibits elastase (made by neutros, which destroys elastin) -- cigarettes and pneumonia inc neutros in lung, thus inc elastase production
- see PiSZ, PiSS, or PiMZ (AAT deficiency assoc c Z allele, done by isoelectric focusing)
MC: PiMM (normal); piZZ causes emphysema, worst px
Misfolded gene product protein accumulates in hepatocyte ER
- Autosomal codominant dz from mutation in SERPINA1 gene cr 14
- dec elastic tissue in lungs --> panacinar emphysema
- causes both lung and liver problems, esp in younger pts
Micro: periportal PAS/D+ inclusions
- cholestatic and hepatic changes
- infants <3 mo may not have the PAS globules, and are best dx'd by serology
Dx: SPEP, CXR, PFT, AAT
- see PAS (+) globules in liber
DDx: megamitochondria (which are randomly distributed, vs periportal distribution in a1-AT def)
- globular amyloid, fibrin globules of fibrinogen storage dz and globules in chronic congestive liver dz can also mimic
Tx: admin of anti-protease, called augmentation tx
- transplant in liver failure
- cigarette smoking will dec lifespan by 20 yrs
Wilson's disease
- aka hepatolenticular degeneration
AR mutation of ATP7B (a transmembrane copper receptor) cr 13 causing : 1) dec Cu secretion into bile; 2) dec Cu incorp c ceruloplasmin; 3) dec ceruloplasmin secretion in blood
- ceruloplasmin accounts for bound Cu fraction
-- see dec serum ceruloplasmin and Cu; inc urine Cu
The binding of Cu to apoceruloplasmic transforms apoceruloplasmin to ceruloplasmin. ATP7B codes for a P-type (cation transport enzyme) ATPase that transports copper into bile and incorporates it into apoceruloplasmin, so absent ATP7B means that copper does not bind, the transformation does not occur, and thus lower levels of ceruloplasmin in the blood
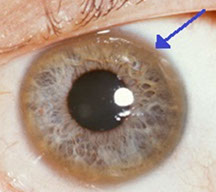
Cu accumulated in liver, brain, cornea (Decemet's membrane), kineys and joints
- usually presents bwt ages of 5-35 yo but can be later or earlier
Sx: ***ABCD: Asterixis, Basal ganglion degeneration (parkinson-like); Cirrhosis, Corneal deposits (Keyser-Fleischer rings), Cu accumulation, Carcinoma (HCC), dec Ceruloplasmin; Dementia; Hemolytic anemia; Fanconi anemia ****
Micro: usually bx'd at later stage of disease, see hepatocyte ballooning, cholestasis, apoptosis in acute stage and macrovesicular steatosis, vacuolization of hepatocyte nuclei and hepatocyte necrosis in chronic dz
- findings can range from almost normal, acute hepatitis, fatty liver dz, cryptogenic cirrhosis
- positive copper stains are usually in a zone 1 distribution, but can be panlobular (not specific finding)
EM: funny mitochondria and fish mouth canaliculi
Dx: inc 24 hour urinary Cu excretion and inc Cu in blood
- dec ceruloplasmin --> inc Free Cu BUT Dec Total Cu
-- bc ceruloplasmin is an acute phase reactant, can be normal in pts c Wilson's dz that have ongoing liver inflam
- sequencing can show the gene mutation in 9/10 c the dz
Tx: copper chelation c penicillamine, possible liver transplant
Px: fatal if untreated; inc risk of HCC
Glycogen Storage Diseases (GSD)
Inc glucose storage in hepatocytes; mostly types Ia/b, IIIa/b, VI, IX, XI
- have patterns of glycogenosis, steatosis, or mix of the two
- PAS stain can show inc glycogen, though not specific
- specific subtype of storage dz can't be made on histology alone, need to correlate c history and clinical findings
- usually present c hepatomegaly, hypoglycemia, recurrent infx, short stature
-- types II and IV not usually assoc c hypoglycemia
-- types III and IV usually have fibrosis (also seen in I and IX)
GSD type 0
Pts present c fasting hypoglycemia and Hmegaly in first year of life
- liver bx c no glycogenosis (glycogen may actually be dec) and macrovesicular steatosis
GSD type Ia/b
Pts present c fasting hypoglycemia and Hmegaly in first year of life
- some can have severe neutopenia and severe inflam bowel dz
- most pts c short stature
Micro: mixed glycogenosis and macrovesicular steatosis
- hepatocytes can have prominent glycogenated nuclei
-- steatosis may be more in young adults
- rarely see fibrosis
- may have hepatic adenomas at time of puberty c inc risk of malig transformation, and are also assoc c anemia that is cured c resection
GSD II
aka Pompe dz or acid maltase def
- have marked cardiomegaly, big tongue, weakness (can have floppy baby syndrome)
Micro: marked glycoenosis, no fibrosis
GSD III
MC in liver and muscle, have Hmegaly, hypoglycemia, and short stature
Micro: marked glycogenosis, fibrosis can be present and progress to cirrhosis (puts pts at risk of HCC)
GSD IV
Andersen dz, branched dec, amylopectinosis
- pts normal at birth, but then fail to thrive and c Hmegaly
- hypoglycemia uncommon
- some can have classic hepatic form, c cirrhosis in early childhood (at ~ 5 yo), though some are not progressive
Micro: ground glass-type inclusions (should exclude HBV and drugs)
GSD VI
Hers dz, fail to thrive, Hmegaly, hypoglycemia
- b9 clinical course c regression of sx as pt matures
- may have focal nodular hyperplasia or adenomas
GSD IX
fail to thrive, Hmegaly, hypoglycemia
- b9 clinical course c regression of sx as pt matures, though a some may have fibrosis or cirrhosis
- marked glycogenosis which is diastase sensitive
GSD XI
Fanconi-Bickel syndrome
- pts c hypoglycemia c post-prandial hyperglycemia, c moon face, fat deposits on shoulders and abdomen
- kidneys involved c prox tubule dysfunction and pts get rickets
- have macrovesicular steatosis and glycogenosis
Lafora Disease
AR, pts c overly branched glycogen molecules which are poorly soluble and precipitate as polyglucosan bodies in hepatocytes
- presents in late childhood, assoc c epilepsy, and is fatal
Urea Cycle Defects
Nitrogen from protein metabolism converted to urea for excretion in urine; defects in this process can cause inc ammonia in tissues
- usually presents in childhood, may be a cause of SIDS, though can present as vomiting, lethargy, irritability, avoidance of meat, hyperactivity, avoidance of high-protein meals
Micro: liver bx c glycogen accumulation, sometimes mild nodular regenerative hyperplasia, megamitochondria and glycogenated nuclei in severe cases
- findings are similar to glycogenic hepatopathy
Mucopolysaccharidoses
Usually AR, rarely X-linked; can cause inc mucopolysaccharides in liver and other tissues
- major dz's are Hunter syndrome, Hurler syndrome, Morquio syndrome, Sanfilippo syndrome, Maroteaux-Lamy syndrome
- micro may show rarified cytoplasm similar to glycogenosis, but will be PAS neg; may be highlighted by iron stains; special fixative soln may be needed to not wash out mucopolysaccharides
Diabetes Mellitus in the Liver
3 main patterns of injury:
1) Glycogenic Hepatopathy
- disruption in the balance bwt glycogenesis and glycogenolysis in hepatocytes 2/2 poor control off blood sugars, causing excess glycogen to build up in hepatocyte cytoplasm
- occurs in pts c type 1 DM c poor glycemic control
- may be part of Mauriac syndrome (rare nowadays bc of better dx and glucose control, but pts c growth retardation, delayed puberty, cushingoid features, hypercholesterolemia, Hmegaly, abnormal liver enzymes, glycogenic hepatopathy)
- pts may get ascites from compression of the sinusoids by rapidly expanding hepatocyte cytoplasm, resolves c better glucose control
Micro: hepatocytes c lots of pale cytoplasm c accentuation of hepatocyte membranes
- PAS can highlight glycogen in the hepatocyte cytoplasm, which is sensitive to diastase
DDx: medication effect (short-term high-dose steroids), malnutrition, dumping syndrome from fundoplication for GERD, GSD
2) Macrovesicular Steatosis
- fairly common
3) Diabetic Hepatosclerosis
- recently delineated, bx shows dense sinusoidal fibrosis
- can be an independent finding not seen c the other 2 patterns
- pts usually c hx of microangiopathic complications from DM in other organ systems, suggesting this pattern is 2/2 microangiopathic dz
Pregnancy and Liver Disease
Hyperemesis Gravidarum
Intractable nausea and vomiting in first trimester that can cause dehydration
- inc liver enzymes, ALT>AST
- enzyme levels return to normal when vomiting resolves
Intrahepatic Cholestasis of Pregnancy
Occurs in 2nd or 3rd trimester, pts c pruritis (usually in palm/soles and worse at night), inc bilirubin and inc ALT, resolves after delivery
- may be assoc c underlying liver dz (HCV, gallstones, PBC, pancreatitis)
- freq has strong regional variation, up to 1/6 pregs, higher in twin preg
- micro: bland lobular cholestasis
Preeclampsia / eclampsia
3rd trimester, <1/10 pregs, HTN c proteinuria +/- HELLP syndrome
- risk factors: fam hx, APL abs, HTN, DM, obesity, twin preg, maternal age >40 yo
- up to 1/3 can present postpartum
Micro: periportal bleeding and fibrin deposits; 1/4 can have microvesicular steatosis; severe cases can undergo infarction
- may have fibrin thrombi in portal vessels and periportal sinusoids (can also be seen in HELLP syndrome)
Acute Fatty Liver of Pregnancy
3rd trimester, fairly rare (though can occur in ~1/10 pregs c triplets), pts c nausea, vomiting, abdom pain, HTN in 1/2
- may be 2/2 defective beta-oxidation of fatty acids
- pts c this dz should undergo testing for the long-chain 3-hydroxyacyl-CoA dehydrogenase (HADHA) gene bc baby can develop metabolic crisis that can be fatal
Micro: diffuse steatosis, predominantly microvesicular, more severe in acinar zone 3 than zone 1, normal hepatic architecture is preserved, portal tracts are unremarkable
Tx: rapid delivery
Normal Canals of Hering staining with CK19. No hepatocytes are positive, indicating there is NOT cholestasis

Hepatocytes stain positive for CK7 in cholestasis



nodular regenerative hyperplasia with regenerative nodule (white arrow) bordered by irregular aligned small-sized hepatic trabeculae (black arrows)

nodular appearance of liver parenchyma (white arrows) characteristic of nodular regenerative hyperplasia. Areas with sinusoidal congestion are also present (black arrow




A1 antitrypsin deficiency in which the synthesized protein lacks the ability to migrate from the ER to Golgi zone and thus accumulates inside the ER as hyaline globules (arrow)




Normal absorption and distribution of copper. Cu = Copper; CP = ceruloplasmin; green = ATP7B carrying copper

Wilson disease, rhodanine copper stain


Funny mitochondria and fish mouth canaliculi in Wilson's


Rhodamine stains, Wilson's
Acute fatty liver of pregnancy. Microvesicular steatosis and ballooning hepatocyte degeneration. Note the involvement of pericentral hepatocytes (left half) with relative sparing of the periportal hepatocytes (right half)

Nutmeg Liver
Liver looks mottled due to bloody congestion in the liver caused by rt-heart failure and Budd-Chiari syndrome
- if persists can cause centrilobular congestion and necrosis resulting in cardiac cirrhosis
Alcoholic hepatitis
Seen in constant long-term alcoholics
Micro: Swollen and necrotic hepatocytes (balloon degeneration) c steatosis, Mallory bodies, zone 3 fibrosis and neutrophilic infiltrates
- Mallory bodies: intracytoplasmic eosinophilic inclusion bodies, stain c intermediate filament, CK8/18, p62, and ubiquitin; in hepatocytes caused by ubiquinated cytokeratin intermediate filaments
Dx: AST/ALT > 1.5, inc GGT (same as in acute viral hepatitis), dec albumin and inc globulins; prolonged clotting times
*** A Scotch and Tonic ***
Alcoholic cirrhosis
Caused by recurrent bouts of alcoholic hepatitis, this is the final, irreversible form of liver dz
- liver is micronodular and small from scarring
Sx: Jaundice, edema (hypoalbuminemia)
Micro: sclerosis around central vein (zone III); Councilman bodies (necrotic liver cells); bridging fibrous septa linking portal tracts, fibrosis, parenchymal nodules
Acute and Chronic Viral Hepatitis
Grading and Staging Chronic Hepatitis
- commonly asked to dtermine amt of inflam (grade) and fibrosis (stage) in pt c known chronic viral hep C or B
- first used scale was Knodell score (scored based on portal tract inflam, interface bridging / panacinar necrosis ), hepatic lobules and fibrosis
-- was found that combo of inflam and fibrosis suboptimal
- later came Scheuer system, Batts and Ludwig system, Ishak system, Metavir system
- Metavir and Ishak used in research (the two can be interconverted)
Inflammatory grade given by adding scores for portal tract inflam, interface activity, lobular inflam
- Metavir uses only interface and lobular activity
- as portal inflam inc, amt of interface activity inc (lobular inflam less closely linked)
- should state the system that you are using to grade if a grade is given in a formal system
Risk factors for progression
older age at first infx, fibrosis on bx, males, infx duration, HIV/HBV co-infx and other liver dz (fatty liver from metabolic syndrome or EtOH liver dz), iron overload
- not a linear progression of fibrosis, inc more rapidly at adv stages
Hepatitis A
Single stranded RNA picornavirus, transmitted via oral-fecal route, possibly by sexual or blood borne also
- vaccine has been available siince 1990
- virus is stable at room temp and resistant to acidic envt
- assoc c eating raw seafood
- incubation perior of 2-7 wks
- 1/3 of infected kiddos have sx, while 4/5 infected adults get sx
- can cause fulminant or fatal hepatitis if co-infx c hep C or B occurs
- does not cause chronic hepatitis, but can recur in transplants from pts c fulminant hep A
- usually self-limited
- can be relapsing and cause prolonged cholestasis
Bx rarely performed in pt c hep A; dx made by serology
Micro: lymphocytic hepatitis c variable lobular and portal inflam, esp portal, which can have inc plasma cells (mimicking autoimmune hep)
- lobular hepatitis zone 3 sometimes
- may see cholestatic lobules and prolif of bile ducts
- cannot tell hep A from other causes of acute hep
- fibrosis not a part of hep A
Dx: + hepatitis IgM HAV abs or PCR for hepatitis A RNA
Px: Self-limiting, give supportive tx
Hepatitis B Virus
partially double-stranded DNA virus
- can be present in high levels in many body fluids; usually transmitted by sex or body fluid/blood exchange
- active hep B has many outcomes depending on age of infx
-- neonates have up to 9/10 of getting chronic infx; adults 1/20
Hep B status divided into immunotolerant, chronic, inactive surface Ag carrier, or resolved infx
- occult infx defined as HBsAg undetectable but DNA levels + in blood or liver
- HBV status and ALT levels do not correlated well with histologic findings
-- although, immunotolerant pts usually have little or mild inflam and fibrosis
Acute Hepatitis B
Rarely bx'd bc dx made c serology and serum PCR for HBV nucleic acids
Micro: portal tracts show lymphocytic infiltrates
- lobules c mod to marked inc lymphs, hepatocyte swelling and occasional apoptotic bodies
- Kupffer cells usually prominent
- lobules can be cholestatic c severe infx
- ground glass inclusions NOT seen (only in chronic hepatitis)
IHC: HBsAg usually negative or only focally pos in acute infx
Chronic hepatitis B
MC reason for bx is stage grade inflam and stage fibrosis
Portal tract changes
Usually has mild to mod portal chronic inflam (lymphs), up to 1/5 c lymph aggs
- portal tract inflam can involve lobules and disrupt row of hepatocytes adjacent to portal tract (called interface activity; previously periportal hepatitis or piecemeal necrosis [term not used anymore bc mechanism of cell death in interface hepatitis is apoptosis and not necrosis])
-- interface activity nonspecific, can be caused by drugs or autoimmune dz, and generally is correlated c degree of inflam
- 1/10 have mild chron inflam of bile ducts and epithelial reactive changes (Poulsen lesion)
Lobular changes
- mild to mod chronic inflam can be seen in lobules in most cases
- marked lobular hepatitis seen c HBV flare or HDV superinfection, but will still be mostly lymphs
- sand glass nuclei are HBcAg nuclear inclusions, assoc c high viral replication levels and are HBcAg+ by IHC; though can also be seen c HDV and are sometimes an incidental finding
-- the HBcAg IHC stain can be pos in cells w/o sand glass nuclei, and will sometimes stain cytoplasm (possibly depending on HBeAg status) and nuclei of bile duct epithelium
- ground glass inclusions can be seen in the cytoplasm of hepatocytes, and are more common than sanded glass nuclei, and are caused by infx of the smooth endoplasmic reticulum (may prevent the cell from releasing viral particles from molecular mutations
-- ground class inclusions may be PAS+
-- HBsAg can stain ground glass hepatocytes and cells w/o inclusions
-- ground glass hepatocytes can be type 1 or 2; but again, ground glass inclusions not specific for HBV (drugs cause pseudoground glass changes)
- flares can cause inc AST/ALT and see mod to marked active lobular hepatitis, with zone 3 foci of necrosis (not usually biopsied)
Granulomas and HBV
~1/50 bx's for HBV have small epithelioid granulomas without polarizable material
- workups are usually negative, though should still do AFB and GMS
Liver Cell Dysplasia
Can be classified as large cell or small cell change / dysplasia and have been assoc c inc risk of HCC (though not necessarily of px significance)
- more common in HBV than other chronic liver dz, but still not entirely specific (also seen c advanced fibrosis)
-- small cell dysplasia is small aggs of hepatocytes c scant cytoplasm but otherwise normal nuclei and cytoplasm
-- large cell change is aggs of hepatocytes c normal to inc amt of cytoplasm but with nuclear hyperchromasia, pleomorphism, and multinucleation
Hepatocyte Oncocytosis
Nodules of oncocytic hepatocytes; not specific for HBV
- unclear significance
IHC: not routinely done, only when labs are equivocal
- the staining patterns may correlate with the clinical category of dz and degree of inflam
-- immunotolerant phase: little lobular inflam, inc nuclear HBcAg in hepatocytes, strong membranous HBsAg
- as inflam inc, nuclear HBcAg decreases and HBcAg cytoplasmic staining inc, with dec in HBsAg
-- carrier state: has distinct circumscribed aggs of HBsAg+ hepatocytes
Fibrosing Cholestatic HBV
Rare form of chronic HBV infx seen in immunosuppressed pts, usually c solid organ transplants and high levels of immunosuppression
- represent the end of a spectrum of dz, and may find other HBV dz findings
Micro: marked lobular cholestasis c hepatocyte swelling or balloon degeneration, mod ductular prolif in portal tracts, and pericellular and portal fibrosis on trichrome
-- the ductular prolif usually suggests obstructive biliary tract dz, and should r/o biliary tract obstruction
-- periportal areas usually c more pericellular fibrosis
- inflam usually mild
- viral levels usually very elevated above baseline, and dz caused by high virus replication levels and direct viral toxicity
- can rapidly lead to fibrosis
Hepatitis D Virus (HDV)
RNA virus that only infects hepatocytes already infected c HBV
- HDV uses the viral coat made by HBV to pack its own nucleic acids
-- can occur as co-infx c HBV, or as a super-infection on HBV
--- super-infection tends to do worse, and are more likely to get chronic HDV, inc risk of worsening fibrosis, and possibly HCC
--- those c co-infx usually clear the infx
- perinatal transmission of HDV is rare, usually acquired through sexual or parenteral routes
Micro: nothing unique for HDV; usually mod to marked acute lobular hepatitis c confluent or bridging necrosis and the normal findings of HBV though usually worse
Labs: superinfection c HDV causes inc liver enzymes without rise in HBV viral DNA
- dx usually made by testing for serum HDV RNA; IHC usually not available but may be helpful
- ELISA or RIA testing for HDAg avail at reference labs
Hepatitis C Virus
Virus is unstable bc of low fidelity of HCV RNA polymerase, causing formation of multiple genotypes and subtypes, called quasispecies
- for this it is hard to develop an HCV vaccine, bc there are dozens of quasispecies in an individual at a time
Acute HCV
Rarely bx'd bc acute infx usually has no or mild clinical sx
- can occasionally be bx'd in elderly
- Micro: cholestatic acute hepatitis c mod to marked lobular inflam and mod lubular cholestasis
- mild to mod chronic inflam (lymphs) of portal tracts
- may see bile ductule prolif c mixed portal inflam in some cases of marked lobular hepatitis
Chronic HCV
Adults and kiddos usually have similar bx findings
- kiddos may have advanced fibrosis despite short time of infx
Portal Tract Findings
Mild or mod lymphocytic inflam in portal tracts and lobules
- usually see somewhat diffuse portal chronic inflam which is at least minimal in all portal tracts and can be mod or marked in medium and large portal tracts
- can have lymph aggs in portal tracts, even c germinal centers (should not be over-interpreted, and does not have much significance)
- interface activity common, which correlates c degree of portal chronic inflam (not useful to try to differentiate "chronic active hepatitis" and "chronic persistent hepatitis", part of older classification system)
-- interface activity does not reflect immune activity against virally infected hepatocytes, bc hepatocytes can be infected when not localized to portal/lobular interface
- plasma cells may be a part of the chronic inflam, may be assoc c low-level Ab titers; and autoimmune hepatitis can occur c chronic HCV
Poulsen (-Christoffersen) lesions - Bile ducts can have mild lymphocytosis and reactive epithelial changes or damage and a prominent lymph agg
- bile duct changes can be assoc c mildly higher ALP or GGT levels, more portal inflam, lymph aggs and more advanced fibrosis
- still not entirely specific for HCV
~2% of explanted livers show bile duct dysplasia
- usually in med-sized bile ducts that are not usually sample on needle bx, but can be on wedge bx or bigger resections
- chronic HCV cirrhosis is a risk factor for intrahepatic cholangiocarcinoma, and dysplasia may be a precursor lesion
Lobular Findings
Mild or mod chronic inflam, rarely severe (which is assoc c flare)
- chronic HCV should not show lobular cholestasis, and should suspect another acute cause of liver injury if found
Giant cell transformation of zone 3 hepatocytes may be seen, and may be assoc c chronic HCV c active injection drug use (not specific)
- may also be seen c HIV co-infection
- not assoc c cholestasis, degree of inflam, or degree of fibrosis
HCV genotype 3 and metabolic syndrome particularly likely to have steatosis ( not specific)
- hepatocyte small and large cell change can be seen rarely
- chronic HCV and HBV may both show chronic venulitis, which is usually mild
- endothelialitis may correlate c degree of overall hepatitis, but unclear
IHC: has not been useful for HCV to date
Granulomas and HCV
1/5 chronic HCV bx's have lipogranulomas
- likely 2/2 mineral oil or lipid droplets released from hepatocytes in setting of fatty liver dz
-- mineral oil is common food additive
- may be assoc c focal fibrosis, but usually an incidental finding
Epithelioid granulomas seen in ~1% of livers bx'd for HCV staging and grading
- usually small and in the portal tracts
- should do AFB and GMS
- significance is unclear
Fibrosing Cholestatic HCV
Rare form of chronic HCV MC in pts c liver transplants, immunosuppression, and high viral replication levels (viral RNA >30 million)
- commonly seen in first year after liver transplant
- histology is similar to that in fibrosing cholestatic HBV (above)
- liver c marked lobular cholestasis c hepatocyte swelling, ductular prolif in portal tracts, and pericellular and portal fibrosis on trichrome
- pericellular fibrosis usually more prominent in periportal areas
(should r/o obstruction by imaging)
- portal tracts sometimes c neuts, lobules c mod to marked cholestasis c hepatocyte swelling and zone 1 / 3 pericellular fibrosis
4 elements in the histologic pattern: cholestasis, hepatocyte swelling, ductular prolif and fibrosis)
- inflam is usually mild, liver injury probably caused by direct viral toxicity
- also represents the end of a spectrum of findings
HCV and Autoimmune Hepatitis Overlap Syndrome
Rarely pts have both HCV and autoimmune hepatitis
- can cause diagnostic confusion
- has no specific findings, though portal inflam is usually greated, and there is more prominent portal plasma cells, and higher grades of lobular hepatitis
- can rarely see marked lobular hepatitis and bridging necrosis or panacinar necrosis (not usually seen c HCV by itself)
Labs: smooth muscle antibodies with antiactin specificity combined with ANA positivity is most helpful
- low levels of these antibodies can be seen in the general population and in pts c nonspecific liver dz
- the presence of antibodies alone do not correlate c inc risk of fibrosis progression or interfere c antiviral tx
"Hepatitis with autoimmune features" is not a distinct histologic or clinical entity
- pts c both these features usually female, older, inc inflam, mildly prominent plasma cells, and more fibrosis on bx
Interleukin 28b (IL28b) Genotype
Predicts which pts will respond to interferon-based antiviral tx for HCV
- genetic polymorphisms (single nucleotide polymorphisms or SNPs) assayed in pts DNA to asses genotype
- CC genotype has much better spontaneous clearance rate and better response to interferon-based tx than TT genotype; the CT genotype has intermediate response
- explains the clinical finding that blacks do not respond as well as whites
Hepatitis E Virus (HEV)
Small RNA virus that causes both sporadic and epidemic dz in developing countries, usually as a waterborn illness
- preg women are at the highest risk for fatality (up to 1/20)
- genotypes 1 and 2 are in deleloping world, and genotype 3 (autochthonous) seen in developed world
- 1/5 US adults have been exposed to HEV by serology, possibly from undercooked pork or wild game consumption
- older adults at highest risk of having clinical dz
Acute HEV
Not really described, but can have variably inflamed, lobular predominant lymphocytic hepatitic, and has some cholestasis
- wide range of findings depending on dz severity
- may mimic drug effect
- a clue may be neutrophils in the sinusoidal infiltrates (unusual for other causes of acute hepatitis); but should but the whole picture together: older pt c acute hepatitis, cholestatic lobular hepatitis, with possible lobular neutrophils
- IHC is not available
- Labs: IgM can be found for 3-12 weeks after start of liver injury, and IgG for a long time
Chronic HEV
Usually in immunosuppressed, esp c organ transplants or receiving chemo
- bx can closely mimic chronic HCV
- a clue may be that chronic HCV usually manifests after 9 months after organ transplant, whereas HEV occurs after several weeks, though there is considerable overlap
Alcoholic steatohepatitis with (1) steatosis (>5%), inflammation, and hepatocyte injury. This image shows all classic features of alcoholic steatohepatitis in a patient with a history of alcohol abuse. Macrosteatosis is obvious and two ballooned hepatocytes are shown containing Mallory-Denk bodies (top and bottom arrows). The arrowhead shows hepatocellular inflammation.
https://www.pathpedia.com/education/eatlas/histopathology/liver_and_bile_ducts/alcoholic_steatohepatitis.aspx


Hepatitis A


Hepatitis B virus





ground glass hepatocyte - hep B, made of HBSAg

Fibrosing Cholestatic Hepatitis

Hep D virus

Hep C virus

Other Viral Hepatidities
Cytomegalovirus (CMV)
Relatively rare, almost always seen in immunosuppressed
- usually mild bx findings, c mild chronic portal and lobular inflam
- viral inclusions not always seen, may need IHC
- may see "mini-microabscesses" c small clusters of neutrophils, which is not specific
Heroes Simplex Virus (HSV)
Also relatively rare, almost always seen in immunosuppressed
- usually see punched-out necrosis, which are circumscribed areas of hepatocyte necrosis, which is variable in size
- overall px depends on the size of necrosis
- multinucleated hepatocytes can be seen
Epstein-Barr Virus (EBV)
Relatively rare, almost always seen in immunosuppressed. or in seemingly healthy adults
- see lobular predominant pattern of hepatitis c lots of lymphs in the sinusoids, though typically few or no acidophil bodies, and the degree of hepatocyte injury is low compared to amt of lobular hepatitis
- hepatocytes can be "beaded" because are large and more active and lined by rows of lymps
- may see small epithelioid and fibrin ring granulomas
Adenovirus (ADV)
Very rare, almost always seen in immunosuppressed, infx is usually fatal;
- no specific pattern, but there is variable hepatocyte necrosis
- hepatocytes at the edge of necrotic regions can have viral cytopathic changes c enlarged nuclei c dark purple smudgy nuclei and can have fatty changes
- IHC can be done to confirm
Echovirus
- enteric virus that usually causes dz in kiddos; infants have high mortality rate
- defining feature is diffuse hemoorhagic necrosis of the liver and adrenals
- the virus targets endothelial cells and can veno-occlusive dz pattern
EBV hepatitis

Adult Giant Cell Hepatitis
- aka postinfantile giant cell hepatitis or syncytial giant cell hepatitis
Pattern of injury that can be seen c multiple etiologies
-- infx suspected port-transplant and can cause progressive fibrosis
- may be linked to HHV-6A, CMV, HEV, EBV
- in HHV-6A the bile ducts can also have giant cell transformation
2 main categories of histologic findings
1) mod to marked giant cell transformation c mild to mod lymph inflam in portal tracts and lobules and lobular cholestasis
- is the classic form of adults giant cell hepatitis and should prompt the ddx of autoimmune hepatitis, drug effect and viral hepatitis
- can be seen c acute liver failure and progression of fibrosis (including cirrhosis)
2) can be seen in chronic HCV c mild but persistent giant cell transformation of zone 3 hepatocytes
- not assoc c inflam grade or fibrous stage, but usually seen on subsequent bx
- different causes of cholestasis can also cause giant cell change (term "giant cell transformation" may be preferred)
Giant cell hepatitis. Multinucleated giant cells seen along with Kupffer cell hyperplasia, suggestive of giant cell hepatitis.[3]

Spontaneous Bacterial Peritonitis
Infx of ascitis c bowel perforation
- MCC: E coli
Dx: Ascites c >250 neutrophils
- fluid should be sent for gram stain and culture
Tx: cefotaxime/ceftriaxone
- can prevent c norfloxin (quinolone) when dec ascites albumin
Other Infections of the Liver
Malaria
Rarely seen as a new infx in developed countries; findings can be mild even infatal cases
- see mild sinusoidal congestion and Kupffer cell hyperplasia c brown-black malarial pigment in the sinusoidal Jupffer cells and / or portal tract macrophages
- do not usually see malarial organisms
- may see jaundice clinically, reflected in lobular cholestasis
- inflam can be absent to moderate
Tick-born Diseases
Ticks can transmit protozoa, bacteria and ciruses, but esp bacteria can elevate liver enzymes
- not too sure about changes seen since liver not usually bx'd in these dz's
Rocky Mountain Spotted Fever
Cuased by Rickettsia rickettsii, transmitted by the wood tick and dog tick; usually infects endothelial cells throughout the body; usually causing GI sx (nausea, vomiting, diarrhea, anorexia) c classic finding of fever, headache, rash and hx of tick bite
- commonly see inc liver enzymnes and hepatomegaly
- may see portal tract inflam c lymphs and neuts, as well as a portal vein vasculitis and fibrin thrombi from organisms in endothelium
- can develop significant cholestasis
Ehrlichiosis
Caused by Ehrlichia and Anaplasma organisms transmitted by the lone star tick; it is an obligate intracellular bacteria that infects WBCs
- >4/5 cases have liver dysfunction, usually mild, which can show as lobular cholestasis and diffuse Kupffer cell hyperplasia
- inflam typically mild overall
Lyme disease
Carried by spirochete, Borrelia burgdorferi, with clinical sx usually nonspecific GI stuff
- micro is variable but with mild to mod hepatitis pattern, with lobular infiltrates c neuts and lymphs
Hepatic Abscess
Can be amebic, fungal or bacterial, usually bacterial in adults
- risk factors are immunosuppression and chronic biliary disease
- may be assoc c cancer (esp of biliary tract and pancreas)
- MC orgs are streptococcal and Pseudomonas spp, and S aureus in kiddos
- bx shows fibrotic rind of tissue surrounding abscess and sometimes central necrotic tissue
- inflam is mostly lymphs, but can have plasma cells with mixed eos and neuts
- scattered reactive bile ducts common in inflamed and fibrotic tissue
- DDx includes inflammatory pseudotumor (IHC not helpful to differentiate)
- stains for organisms may be helpful
Actinomycosis
Filamentous bacteria that can have hyphal-like structures that mimic a fungal infx; rarely primary to liver, MC from other organs
- infx forms mass-like lesion in liver that can be single or multiple and sometimes mimic liver tumors
- most pts are immunocompetent, and it is MC in males (2M>1F)
- micro shows rind of inflamed fibrotic tissue possibly c central areas of necrosis, and can also mimic an inflammatory pseudotumor
- organisms are GMS+
- usually grow in large colonies that have sulfur granules and appear purple on H&E
- 1/3 of infx are polymicrobial, and different organisms can be seen
Whipple Disease
Caused by Tropheryma whipplei, gram+ organisms seen in PAS+ macrophages, that presents c arthralgia, weight loss, diarrhea and abdominal pain
- liver can be involved in systemic dz, but usually not isolated in liver
- may see nonspecific Kupffer cell hyperplasia
- arteriopatrhy can be seen, and organisms in the arterial wall can be seen in PAS/GMS stains in vessel walls
Echinococcis
Hydatid disease caused by larval form of Echinococcus tapeworm
- MC organisms are E granulosus and multiocularis
- sheep are MC intermediate host, and life cycle completed when viscera of infected sheep are eaten by dogs, which are the MC definitive host
- humans are an intermediate host and become infected by exposure to infected dogs or contaminated water/food
- parasitic cysts can involve brain, heart, kidney lung and spleen
- liver cysts are slow-growing and can reach large sizes
- most cases dx'd by imaging c serologic confirmation
- bx not usually performed by ruptured cysts can cause ana[hlyactic shock
The hydatid cyst has three histologic layers c outer pericyst and germinal layers
- cyst fluid can be hemorrhagic and grungy-appearing on H&E, usually c dead organisms
Autoimmune Hepatitis (AIH)
Chronic, self-perpetuating, immune-mediated liver damage
- variable over time, middle-aged women, Hyper-IgG
- to dx, must r/o viral hepatitis and drug reactions and must see histologic features on bx
4F > 1M, though M=F in prepubertal and elderly pts
- bimodal age distribution, first in 10-20 yo then ~40 yo, though 1/5 occur after 60 yo; strong assoc c DRB1* alleles
- type 1 AIH assoc c HLA DR3 and HLA DR4, though the mechanism not understood; assoc c ANA, SMA, anti-SLA/LP and sometimes AMA
- type 2 AIH in kiddos and teens, can be anti-LKM-1 (against CYP2D6) or ACL-1 abs
- presents as unexplained hepatitis in 1/4 of pts, and 1/2 have non-specific sx like fatigue, malaise, abd pain
- 1/4 are asx
- the Intl' AIH Group made an early scoring system, in which the pathologists' goal is to determine if the histology is compatible c AIH
"Typical" AIH
1) interface hepatitis, c lymph/plasmacytic infiltrates in portal tracts extending to lobule
2) Emperipolesis, c active penetration by one cell into another cell
3) Hepatic rosette formation
- basically any chronic lymphocytic hepatitis is sufficient to say "compatible with AIH"
Dx: serum IgG levels typically inc
- positive for abs: antinuclear antibodies (ANA), antismooth muscle antibodies (ASMA), anti-liver/kidney microsomal (LKM) antibodies, liver cytosol type 1 antibodies (LC-1), and soluble liver antigen (SLA)
- antibodies used to define the subtype of AIH
Type 1: ANA, SMA abs, Female <40, most common
Type 2: LKM1 (liver-kidney microsome 1) abs, children 2-14
Type 3: SLA/LP (soluble liver antigen/liver pancreas) abs, Female <40
Hypergammaglobulinemia, no cholestasis
Only chronic hepatitis that responds to corticosteroid
Type 1 AIH - MC after puberty
- 19/20 cases of AIH, 1/2 have both ASMA and ANA positivity, 1/3 c just ASMA +
- very increased serum IgG levels
- 1/2 go to cirrhosis
- Micro: hepatitis c prominent plasma cells
Type 2 AIH - also seen in kiddos
- abs tend to be at lower titers in the peds population
- have LKM type 1 and LC-1 ab +
- only slightly elevated IgG levels
- 4/5 progress to cirrhosis
- Micro: hepatitis c prominent plasma cells
Many self-ags are the targets of ANA, such as ds-DNA, chromatin, and ribonucleoprotein
- ASMA targets filamentous actin (F-actin), vimentin, and desmin (ASMA against F-actin is the most specific for AIH)
- LKM-1 abs are against cytochrome P450 CYP2D6
- LC-1 abs are against formiminotransferase cyclodeaminase
- anti-SLA considered to be the same ab as anti-liver/pancreas (LP) and is against selenocysteine synthase
Must be careful bc lots of inflam conditions can have low positive titers
- high titer auto-abs can be seen in pts c mild liver enzyme inc but no significant hepatitis on bx, which is insufficient for a dx of AIH
Micro: typical pattern in acute AIH is mod to marked portal inflam c prominent plasma cells, interface activity and mod to marked lobular hepatitis, confluent necrosis
- if there is no confluent necrosis, nearly impossible to have "severe activity"
- lobules also have occasional acidophil bodies, var hepatic ballooning and lobular disarray
- severe cases c lobular cholestasis and zone 3 necrosis
- must remember that this pattern variable, and prominent plasma cells can be found in viral and drug-assoc hepatitis
- the lobular hepatitis can have lobular cholestasis and hepatocyte rosettes if marked; lobular neuts are uncommon and suggest other dz
- fibrosis does not matter to dx AIH, but should be reported
Type 1: ANA, SMA abs, Female <40, most common
Type 2: LKM1 (liver-kidney microsome 1) abs, children 2-14
Type 3: SLA/LP (soluble liver antigen/liver pancreas) abs, Female <40
Hypergammaglobulinemia, no cholestasis
Only chronic hepatitis that responds to corticosteroid
Micro: Very active interface and lobular hepatitis
- Abundant plasma cells in clusters, eosinophils
- Regenerative periportal liver cell rosettes
- Severe bridging necrosis, cirrhosis
If livers c advanced fibrosis are started on immunosuppressive tx, may form regenerative nodules that mimic tumors or imaging studies and gross exam
- nodules can be up to several cm in size, are haphazardly distributed, and have background liver c lots of parenchymal collapse
IHC: may be helpful to distinguish from PBC
- in AIH, IgG-+ plasma cells predominate
- in PBC, IgM + plasma cells predominate
DDx:
- Drug effects - can closely mimic AIH by serology and histology
- Overlap syndrome with PBC and AIH or PSC and AIH
Tx: immunosuppression
- responds to corticosteroids, c/wo azathioprine common
Autoimmune Sclerosing Cholangitis (ASC)
- overlap bwt AIH and PSC, MC in kiddos and young adults
-- autoimmune cholangitis (without "sclerosing") is an AMA-neg PBC
- ASC has typical features of AIH and imaging studies typical of PSC
- UC is strongly linked to both ASC and PSC


Mo to yrs
burned out hepatitis
scar is direct response to activity
<- little scarring, severe activity
Autoimmune hepatitis, lymphocplasmacytic infiltrate with interface hepatitis


Severe activity with parenchymal collapse showing 2 portal tracts next to each other without intervening hepatocytes


Confluent necrosis - hepatocyte dropout around central vein

Bridging necrosis

Total parenchymal collapse
Confluent necrosis (peri-venular necroinflammatory activity)

Bridging confluent necrosis

AIH with hepatocyte rosettes
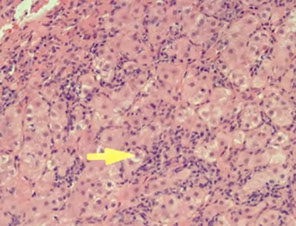
Drug Effects
Common cause of acute hepatitis and prompt the clinician to order a liver biopsy; aka Drug Induced Liver Injury (DILI)
- MC drugs are acetominphen, abx, CNS agents, antihypertensives, and diabetic meds; dietary supplements are another common cause
- the pattern of reaction can vary substantially, and any drug can cause multiple patterns of injury
-- for example, tamoxifen can cause acute hepatitis, massive liver necrosis, peliosis hepatitis, steatosis, steatohepatitis, and cholestasis
- should search reliable source when drug reaction suspected to see published literature for the drug
Basic Mech of Injury
Can be directly toxic, allergic or an idiosyncratic drug reaction
- MCC is idiosyncratic
Making the dx depends on having the hx, compatible histology, laboratory testing results (such as hep E or C), and excluding other dz's
- toxicity of a drug assoc c the amt of intake, and most drug reactions occur in the first several week of exposure to a medication (though can take longer)
Resolving Pattern of Hepatitis
Sometimes a drug reaction suspected and a drug is stopped, though the liver enzymes remain elevated for weeks to months after discontinuing the drug
- bx in this case can show resolving hepatitis or significant ongoing injury that suggests another dx
Resolving hepatitis shows minimal or mild portal lymphocytic inflam, and absent to minimal lobular lymph inflam; lobules show mild Kupffer cell prominence which may have pigmented PASD+ material
- lobules can have mild cholestasis
- generally, hepatocyte injury resolves after stopping a drug, though cholestasis can resolve more slowly
Histologic Clues to a Drug Reaction
Hypersensitivity type drug reaction suggested by prominent eos
- bland lobular cholestasis also seen, which does not have fibrosis or inc inflam (not specific, but commonly seen c drug rxn)
Direct toxins
Cause liver injury in dose-dependent and reproducible way
- includes acetaminophen, mushroom toxicity, misc household and industrial chemicals
-basic injury pattern is direct necrosis c little inflam, with surviving hepatocytes showing fatty change c small- and med- fat dropllets, cholestasis and other reactive changes
-- necrosis usually c zone 3 pattern
-- toxicity from phosphorous, ferrous sulfate and cocaine have zone 1 pattern of necrosis
-- beryllium toxicity assoc c zone 2 pattern of necrosis
-- more severe necrosis has panacinar necrosis without zonation
Acetaminophen Toxicity
Classic example for direct liver toxicity, c the degree of injury proportional to the level of exposure
- causes the majority of acute liver failure in the USA and UK
- usually happens in pts that take large doses unintentionally, but also as a suicide attempt (usually assoc c EtOH use or c chronic pain)
- liver injury starts to happen c 7.5 g of exposure, c severe damage c 15-25 g exposure (though the threshold can be lower in pts c chronic alcohol use, fatty liver dz, and drugs that stimulate the P450 enzyme system [carbamazepine, cimetidine, isoniazid, and phenytoin])
- mediant ALT level c acetaminophen tox is 4300 IU/L, while bilirubin is usually only mildly inc (~4 mg/dL)
- tx c N-acetylcysteine is very effective if given in the first 24 hours
Micro: hepatocyte necrosis c zone 3 distribution in mild cases, with panacinar necrosis in severe cases
- Kupffer cells and endothelial cells usually still alive and seen in their normal locations
- remaining hepatocytes often cholestatic c mild microvesicular steatosis and scanty inflam
- if bx taken a while after the drug consumption, will see a little more inflam in the necrotic areas, as well as areas of parenchymal collapse c bile ductular proliferation
-- in these later bx's proliferating bile ductules and Kupffer cells can have significant iron accumulation
IHC: CD10 or CEA will show loss of the canalicular staining pattern in the necrotic areas
Allergic-type Drug Reactions
Rarely bx'd bc pts usually have characteristic sx such as hives, wheezing and peripheral eosinophilia
- usually the drug exposure is in the prev days or weeks
- pts c chronic viral hepatitis, autoimmune hepatitis, and PBC can also have inc portal eos in the pattern of a drug rxn; inc eos in peripheral blood may cause inc sinusoidal eos
Micro: eos inc in portal tracts and sinusoids
- lymphs can also be mild to mod inc
Idiosyncratic Drug Reactions
Not dose related and can't be predicted on individual basis
- MC type of drug rxn seen on liver bx, but must exclude viral hepatitis and autoimmune hepatitis
- usually not assoc c fibrosis, should consider another injury pattern if extensive fibrosis is seen
- some of the patterns include: hepatitic pattern, granulomatous pattern, cholangitic pattern, cholestatic pattern, ductopenic pattern, fatty pattern
Hepatitic pattern
Has inflam changes that resemble acute hepatitis
- mild to marked lobular inflam c variable lobular disarray and apoptotic hepatocytes; inflam mostly lymphs
- lobular cholestasis seen c severe degrees of lobular hepatitis, as well as zone 3 necrosis
- portal tracts c mostly lymph inflam, c occasional plasma cells, neuts, and eos seen
- interface activity may be present, esp c moderate or marked portal inflam
Differential includes viral and immune hepatitis; must correlate labs/hx
- idiosyncratic drug rxns (esp minocycline and nutrofurantoin) can have lots of plasma cells in a hepatitis pattern, and can also be ANA and SMA +
Granulomatous pattern
Lymph inflam of portal tracts and lobules, and may have granules in the bile ducts
- are usually assoc c mod to marked hepatitic changes
- can be well or poorly formed epithelioid granulomas
- should do the usual granuloma workup
- broad differential, fibrin ring granulomas assoc c allopurinol
Cholangitic pattern
Actuve bile duct injury c lymphs in bile duct epithelium, reactive epithelial changes and apoptotic epithelial bodies
- inflam usually mixed c lymphs and neuts
Cholestatic pattern
MC c oral contraceptives, anabolic steroids, abx, HAART
- usually c zone 3 predominance
- bland lobular cholestasis can be seen for months after drug is stopped, usually does not have much inflam
Ductopenic pattern
Important pattern, may be subtle, CK staining can be helpful in evaluating loss of ducts
- loss of blie ducts in large portal tracts is abnormal, while it may be absent in 50% of smaller portal tracts
- overall pattern may mimic rejection of liver allograft
- ductopenia less likely drug related if there is ductular reaction, bile duct duplication, onion skinning fibrosis, or fibro-obliterative changes
Fatty pattern
Can cause microvesicular and macrovesicular injury pattern
- ~1/50 NAFLD are drug-related
- steroids are some of the MC drug-induced causes of steatosis
Medications assoc c Fibrosis
Uncommon in most drug reactions, though has been assoc c several drugs (may be difficult to prove causlatiy in most cases)
- cirrhosis may be assoc c tamoxifen, ebrotidine, and amox-clavul acid
Methotraxate
used in management of psoriasis and RA, though has a known risk of fibrosis, esp in pts c other chronic liver dz, such as fatty liver dz and EtoH-related dz
- liver bx usually done at baseline and then done periodically to monitor for fibrosis (should also report steatosis and active injury)
Micro: fibrosis can be both portal and pericellular, and can also show fatty changes, hepatocyte nuclear pleomorphism, and mild to mod portal lymph inflam
- fibrosis grading with methotrexate use has its own grading schema
Excess Vitamin A
Recognized cause of liver dz (fibrosis and cirrhosis), usually will not find excess of vit A intake in the pts clinical hx
- MC presentation is mild persistent elevations of AST and ALT and the bx looks "almost normal" at first glance
- serum vitamin A tests may show normal levels, which is misleading bc vit A circulates as esters bound to plasma proteins
Micro: see an increase in enlarged, lipid-laden stellate cells in the sinusoids
- can also see nodular regenerative hyperplasia and mild chronic inflam; with fibrosis setting in years to decades after
- immunostains not really helpful
DDx: cytoplasmic vacuolization in Kupffer cells, usually seen c cholestasis
Vascular changes assoc c drug effects
Chemotherapy can cause sinusoidal obstructive change (aka veno-occlusive disease) and nodular regenerative hyperplasia
Drug-induced cytoplasic changes, including inclusions
Changes caused by smooth ER (endoplasmic reticulum) prolif and can have several different patterns
- most pts are immunosuppressed c multiple medications
- should get immunostains to r/o HBV infx
Micro:
- hepatocytes can have diffuse gray homogenous cytoplasmic change
- also seen in pts c HIV/HCV coinfection as incidental finding
- similar pattern reported c phenobarbitol and barbiturates
- smooth ER prolif can show zonation, usually called "induced" hepatocytes
- another pattern of smooth ER prolif is "two-tone" hepatocytes, where hepatocytes have two colors in their cytoplasm (usually red and gray) and is best seen c PAS
- yet another pattern is glycogen pseudoground glass, which are also smooth ER cytoplasmic inclusions (which can mimic chronic HBV)
Drug reactions can also cause hepatic glycogensosis, with enlarged hepatocytes having pale cytoplasm filled c glycogen
- MCC is corticosteroids (which can also cause steatosis)
for methotrexate liver bx eval


Hypervitaminosis A liver bx c stellate cells

Pseudoground glass in hepatocytes (polyglucosan bodies)

Hepatic glycogenosis

Fatty Liver Disease
Nonalcoholic Fatty Liver Disease (NAFLD)
MC type of fatty liver dz seen in bx
- morphology usually straightforward, though the question arises:
1) how to differentiate from steatohepatitis vs steatosis
2) is the pt has low-titer ANA + and some portal chronic inflam, how to tell if there is a component of autoimmune hepatitis
3) what scoring system should be used
Look for: 1) Steatosis, 2) Steatohepatitis, 3) Steatofibrosis
- should mention all 3 in dx, if bx sent to eval for "NASH"
- fat usually starts to accumulate around zone 3
- in steatofibrosis the central vein is drawn closer to portal tract
Most pts c NAFLD are middle-aged adults, though can be in kids/teens
- AST and ALT are mildly elevated usually, but can be normal
- metabolic syndrome is the most important risk factor for NAFLD
- pts do not drink EtOH, or do so in small quantities
-- another assoc is chronic intermittent hypoxia from sleep apnea
Two-hit model for NAFLD:
1) Insulin resistance causes hepatic steatosis
2) hepatocellular oxidative injury causes liver cell necrosis and inflam
Metabolic syndrome
MCC of NAFLD in Western countries, usually defined as central obesity, dyslipidemia, raised blood pressure and elevated fasting serum glucose levels with insulin resistance
- obviously, not everyone with metabolic syndrome has NAFLD, and vice versa
Steatohepatitis has a greater risk of turning into fibrosis than steatosis by itself
- fibrosis is usually progressive, so minimal fibrosis on an early biopsy often a harbinger of more fibrosis, and possibly even HCC
- the amt of fat seen can decrease as fibrosis progresses
Autoantibodies can be positive in up to 1/3 of pts c NAFLD
- usually ANA or ASMA (anti smooth muscle antibody) +, at low titers and may be assoc c advanced fibrosis and active dz
- NAFLD may also co-occur with autoimmune hepatitis
Micro: core findings are fat, balloon cells, inflam and fibrosis
- Steatosis usually macrovesicular, with large fat vacuoles, one per cytoplasm, which push the nucleus to the side, though can be smaller (the large ones must come from somewhere...) and can be graded from minimal (<5%) to marked (>67%)
-- steatosis can be around central veins (zone 3) or portal tracts (zone 1), though easiest usually to tell where the fat is not
- fnflam generally mild, inc inflam more assoc c fibrosis
-- surgery itself (wedge biopsy) can cause surgical hepatisis
Balloon cells are hepatocytes that are not yet dead, with lots of clear cytoplasmusually c scattered small eosinophilic clumps and maybe a little Mallory hyaline (damaged and ubiquinated cytoskeletal proteins), though they usually do not have fat droplets
- usually seen in zone 3
- can be seen in multiple diseases (cholestatic liver dz)
Lipogranulomas MC in portal tracts or central veins
- made of groups of histiocytes c lipid droplets
- many seen c food additives (mineral oil), but also c NAFLD or HCV
Megamitochondria common in NAFLD, and are small red dots in hepatocyte cytoplasm, may be single or multiple
- nor specific for NAFLD
1/10 cases have small circumscribed patches of microvesicular steatosis, usually seen c marked steatosis c active steatohepatitis
- also common in advanced fibrosis
- similarly may see areas c hepatic glycogenesis
- glycogenated nuclei seen in ~1/2 of steatosis, and may correlate with diabetes
Tx: weight loss, inc exercise, balanced diet, EtOH avaoidance, vit E may help
Steatohepatitis
Micro: Centrilobular (begins in zone 3) injury c Steatosis; Ballooned hepatocytes; Mallory-Denk bodies; Ropey, pink material;
- Intermediate filaments metabolized by cytokeratin from liver cell injury: p62+, ubiquitin+
– Fibrosis (perivenular/pericentral) of zone III
– Inflamed lobules (neutrophils)
– Megamitochondria: Globular, eosinophilic material indicates
oxidative injury to mitochondria
Risk factors: Alcohol, Diabetes, Hypercholesterolemia, Obesity,
Drugs Amiodarone (Foamy granular hepatocytes from phospholipids which show lysosome inclusions by EM)
– Methotrexate
– Tamoxifen
– Naproxen
– TPN
– Trimethoprim Sulfa
Differentiating Steatosis from Steatohepatitis
- Important distinction to make, bc steatosis has little risk of progression to fibrosis, whereas steatohepatitis results in fibrosis in 15% and cirrhosis in 30%
- histologic continuum exists bwt the two
- both diseases should have some fat, and both can have fibrosis, but steatohepatitis also has active or ongoing inflam /injury
-- active injury can have ballon cells (+/- Mallory hyaline), lobular hepatitis, and apoptotic bodies
-- zone 3 pericellular fibrosis may increase your certainty that the process is steatohepatitis
Fibrosis staging
Two patterns of fibrosis in fatty liver dz
1) perisinusoidal fibrosis / chicken-wire fibrosis
- begins in zone 3; may be diffuse, and as progresses can have portal fibrosis or eventually bridging fibrosis or cirrhosis
-2) portal-based fibrosis more common in kiddos c FLD
Scoring systems
Includes the NAFLD Activity Score (NAS), where biopsies with a score of 0-2 usually steatosis, and cores 5 and up usually steatohepatitis
- scores of 3 and 4 can be either
Steatosis, Activity and Fibrosis (SAF) score also used
- uses separation of fat from ongoing hepatic injury
Methotrexate toxicity can cause a steatosis-like rxn, but it is different and has its own grading system
- must suspect in a pt c hx of psoriasis (gets treated c methotrexate)
Alcohol-Associated Fatty Liver Disease (EtOH FLD)
Shares many histologic features c NAFLD
- can have from just steatosis to marked active steatohepatitis
- should look for clinical hx of EtOH abuse
EtOH FLD favored over NAFLD if:
1) diffuse sclerosis in central veins
2) striking balloon cells and Mallory hyaline
- usually also has prominent neuts in the lobules
- trichrome also shows strong and diffuse pericellular fibrosis throughout the lobules
Other causes of Macrovesicular Steatosis
- first should exclude NAFLD, drug effects and EtOH as cause of FLD
Malnutrition
- categorized as either marasmus or kwashiorkor, can be seen in western world in cases of abuse, or by feeding kiddos nonstandard diets
-- kwashiorkor causes by sufficient calorie intake by not enough protein, seen mostly in infants that are being weaned from breast feeding, and >9/10 pts c this kwashiorkor also have FLD
- FLD also common c marasmus, where there is insufficient total calorie intake
Abdominal surgery
- when parts of bowel are removed, and the pt functionally suffers from malnutrition
Portal vein thrombosis
- FLD seen in 4/5 of these pts, mechanism unclear
Wilson disease
- in up to 1/10 kiddos c Wilson disease can be presenting sign and can mimic NAFLD; best to be suspicious if bx from teen or young adult without metabolic syndrome
Microvesicular Steatosis
Distinct injury pattern where hepatocyte cytoplasm filled c lots of small drops of fat that gives hepatocytes a bubbly look
- assoc c mitochondrial injury from various causes and can be seen c drug reaction
Alcoholic Foamy Liver Degeneration
Unique pattern of EtOH-related liver injury where bx shows diffuse microvesicular steatosis
- seen c zone 3 distribution in mild cases
-- megamitochondria may be seen in spared hepatocytes
- usually in chronic alcoholics that go on a binge
Balloon cells
Mallory hyaline


Megamitochondria

Nonalcoholic Steatohepatitis Clinical Research network Fibrosis Staging System


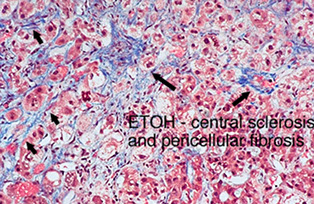

EtOH central vein scleosis
Granulomatous Disease
Granulomas seen in ~4% of liver bx's, MC assoc c primary biliary cirrhosis (PBC causes ~1/2), sarcoidosis (causes ~10%), infx, drug effect, autoimmune dz or idiopathic (~1/3)
- can be part of a bigger dz process that involves other organs, or may just be found in the liver, and the etiology not usually evident by histology alone
- etiology may be narrowed down by certain histologic features
First should decide if the liver dz is a primary granulomatous process (ie sarcoid) or just part of a pattern of injury from another dz (such as in PBC)
- the location of the granuloma usually does not tell much about etiology
Granulomas c large central necrosis are usually infectious
- AFB and GMS should be done on every case, and may be helpful to get second set of stains on deeper sections
- reports for granulomas c large central necrosis should state that infection is suspected even if stains are negative
- sarcoid can cause large central necrosis, though it is usually without the "dirty" nuclear and cellular debris seen c infection
Granulomas c mostly portal granulomas of differing ages (such as old fibrotic granulomas and fat and fresh epithelioid granulomas) is seen c sarcoidosis
Granulomatous biliary tract dz usually from PBC or drug effect
- sarcoidosis also in differential (caused by hilar LN enlargement?)
- parasitic infx of bile ducts also cause granulomas
Drug reaction causes granulomatous dz c mod to marked lobular hepatitis; should just look up meds online to see if they can cause it
- drug reactions and infx can cause granulomatous dz that presents as acute hepatitis
- epithelioid granulomas should be polarized to see if they contain foreign body material
Caseating Granulomas
Have "dirty" central necrosis, c nuclear debris and dead cellular material; MCC is infx
- small areas (a few cells big) c equivocal fibrinoid necrosis not truly caseating most of the time
Epithelioid Granuloma
Aggregate of histiocytes c sharp borders; and are amphophilic to slightly eosinophilic, one of the MC granulomas in liver pathology, should be worked up for bugs
Fibrin Ring Granuloma
Have central fat droplet with surrounding ring of variable thickness and outer layer of macrophages; seen c infx, sometimes drugs
Florid Duct Lesions and Granulomatous Inflam
Medium-sized bile duct cuffed and infiltrated by lymphs
- duct epithelium injured, and has reactive changes
- can be assoc c lots of macrophages c ill-defined aggregates in portal tracts (is also called granulomatous)
- may be assoc c PBC if found in the lobules
Foreign Body Granuloma (FBG)
- many etiologies, such as talc, prior surgery, tumor embolization
- best seen under polarized light
Granulomatous Inflam
Histiocyte-rich inflam, which does not form well-defined granuloma seen in lots of dz's but assoc c drugs and biliary tract dz in portal tracts and drugs or infx if lobular granulomatous inflam
- may be seen if wall of abscess bx'd
Lipogranulomas
Made of small cluster of macrophages c small droplets of lipid or fat
- MC in hep C or fattly liver dz
- classical assoc c mineral oil (food additive)
Microgranuloma
Small group of Kupffer cells in hepatic lobules, caused by repair of an acute episode of hepatitis
- no necrosis or polarizable material
Sarcoidosis
Etiology unknown, may be 2/2 infx, usually <40 yo F, MC in Scandanavians, more aggressive in blacks, possible assoc c autoimmune dz
- have granulomas in different organs, usually lungs, LN and liver
4 main patterns of dz:
1) scattered, mostly portal granulomas of varying age (some c fibrosis)
- can rarely have Schaumann or asteroid bodies (not specific)
2) granulomas plus chronic obstructive biliary tract dz
- caused by enlarged hilar LNs obstructing extrahepatic bile ducts
- can see duct prolif, ductopenia, and portal fibrosis
3) severe portal granulomatous venulitis and loss of portal veins
- lobules may have nodular regenerative hyperplasia
4) rarely can have mass lesions c confluent fibrotic granuloma
- aka sarcoidoma
Other diseases with granulomas:
1) Acute cellular rejection - may disappear after anti-rejection tx (thus may be part of the rejection process)
2) Bacillus Calmette-Guerin - tx common c TCC of blader
3) Celiac disease - can show mild nonspecific inflam changes, and sometimes small portal or lobular epithelioid granulomas
- Celiac dz assoc c inc freq of sarcoidosis and PBC, so you should be primed to look for histologic and clinical features of these dz's
4) Crohn disease - rarely see granulomas, may be difficult to distinguish from mild nonspecific hepatitis, fatty liver dz or primary sclerosing cholangitis
5) Common Variable Immunodeficiency (CVID) - can see mild portal chronic inflam w/p plasma cells and mild to focal moderate lobular lymph inflam; granulomas are small, noncaseating and epithelioid
- lung hilar adenooathy and inc serum ACE levels seen in both CVID and sarcoidosis
6) Idiopathic - 1/3 of granulomas in liver idiopathic despite workup
7) Primary Biliary Cirrhosis (PBC) - granulomas are a well-known part of PBC, which can be epithelioid or as a poorly formedxanthogranulomatous inflam of medium-sized bile ducts
8) Rare causes of granulomas - gout vasculitis, polymyalgia rheumatica, juvenile chronic arthritis, GVHD, bypass surgery, resolving chronic biliary tract dz
- tumors such as Hodgkin disease, HCC, fibrolamellar ca, cholangioca, mets can be assoc c granulomas
Foreign Body Granuloma (FBG)
Polarization can identify foreign material
- common after chemoembolization tx, also in injection drug users
Liver Failure
Acute Liver Failure
Encephalopathy and coagulopathy that manifest w/in 6 months of initial hepatic insult in a pt c no prior hx of liver dz
- caused by massive hepatic necrosis
- usually caused by chemical substances (acetaminophen) if very acute; viruses take a little longer (months)
- clinically divided into: hyperacute (<1 week), acute liver failure (1 week to 1 month), subacute liver failure (1 to 3 months), and must lose >80-90% of liver function
Sx: jaudice/icterus, hepatic encephalopathy, coagulopathy (prothrombin time > 15 sec or INR >=1.5)), portal-HTN, hepato-renal syndrome
Gross: liver is shrunked up
Micro: depends on the etiology;
- toxic / ischemic injury: massic necrosis c mild inflam
- med rxn: marked cholestasis and inflam
- fatty liver of pregnancy, mitochondrial dz: microvesicular steatosis
Survival px correlated with % hepatocyte necrosis (death is rare c <25%, and if >75% need liver transplant or greater risk of death)
Dx: hepatic enzymes get very high (returning back to normal doesn't always mean pt is getting better; conversely could mean all hepatocytes are dead and pt is dying!!)
- ~4/5 die if they do not get a transplant
Chronic liver failure
MCC: Hep B/C, NAFLD, EtOH
- usually (but not always) seen in conjunction c cirrhosis
- Child-Pugh classification: A (well-compensated) to C (decompensated)
Portal HTN: 1) Ascites, 2) portosystemic venous shunts, 3) congestive splenomegaly, 4) hepatic encephalopathy
1) Ascites: fluid in peritoneal cavity caused by sinusoidal HTN (inc vessel pressure and low albumin), drainage of hepatic lymph into peritoneal cavity, splanchnic vasodilation
2) portosystemic venous shunts: produces hemorrhoids and esophageal varices (high mortality)
3) Splenomegaly, may be assoc c pancytopenia
- 1/3 of pts also have hepatopulmonary syndrome, which has a poor px
Cirrhosis
Chronic liver dz c diffuse disruption of hepatic architecture and fibrosis (scarred nodules)
- if nodules >3 mm, think infx or drug-induced hepatitis; if <3 then EtOH, hemochromatosis, or Wilson's dz
Sx: Jaundice (dec bilirubin breakdown), palmar erythema, spider nevi, gynecomastia and testicular atrophy (dec estrogen breakdown), asterixis, anemia, coma and fetor hepaticus (from hyperammonemia), dec platelets, dec LDL, inc PT/INR
- from portal HTN, get ascites / edema, hematemesis, caput medusae, hemorrhoids, splenomegaly
Normal liver has type I / III collagen around portal tracts and type IV in space of Disse
- in cirrhosis, type I/ III collagen are deposited in the space of Disse causes portal tract fibrosis which causes a loss of the fenestrations in the sinusoidal epithelium thus impairing the functions of the sinusoids.
-- the KEY mech of fibrosis is prolif of the stellate (Ito) cells and their transformation to myofibroblasts, which deposit collagen and have contractile properties the cause sinusoidal constriction
--- Kuppfer cells make cytokines that facilitate the Ito --> myofibroblast conversion
Ductular reactions inc with more advanced stages of liver dz and are most prominent in cirrhosis
Dx: SAAG (Serum Albumin: Ascites Gradient) = [serum albumin] - [ascites albumin]
- >1.1 in portal HTN (transudative); <1.1 in cancer, nephrotic syndrome, TB, pancreatitis, biliary dz, CT disease
MCCs death: liver failure, portal HTN (ruptured esophageal varices), hepatic encephalopathy, HCC
Tx: avoid hepatotoxins, B vits and inc dietary protein (for dec albumin), salt restriction and diuretics, transplant in liver failure
- TIPS (Transjugular Intrahepatic Portocaval Shunt) can ameliorate portal HTN
Hepatic Encephalopathy
See in pts c asterixis, confusion, sleep cycle reversal, changes on EEG
- lots of predisposing factors
caused by 1) inc [ammonia] in blood (liver cells can't convert it to urea); 2) production of false neurotransmitters; 3) inc CNS sensitivity to GABA; 4) zinc deficiency
Dx: LFTs
Tx: Lactulose, rifaximin / neomycin
Liver Transplant
Donor Liver Eval
Bx done to see if liver ok to transplant, but decision not only based on bx, also age, warm ischemia time, donor and recipient history
- pathologist should report on amt of fat, inflam, necrosis, fibrosis, other stuff
- fat eval best done c H&E, should report amt macrovesicular steatosis bc if <30% performs as well as a transplant c no fat, and >60% have a poor chance of primary graft nonfunction
-- MICRO-vesicular steatosis does NOT affect outcome
- if chronic portal inflam is more than mild, donor may have undiagnosed chronic hepatitis
- not sure whether inc iron levels assoc c worse px
Allograft Preservation Changes
May vary in severity, but can include hepatocyte apoptosis, mild portal / lobular lymph inflam, scattered lobular Kupffer cell aggregates
- occasional balloon hepatocytes with mild cholestasis can be seen, usually more in zone 3 hepatocytes
- fat lobules released from dying hepatocytes can stay in hepatic cords and mimic dilated sinusoids, and can eventually cause real sinusoidal obstruction and cause fat emboli in the lungs
Acute Cellular Rejection
- an immune mediated, lymph-based inflam response to allograft liver by recipient immune system, usually in first months after transplant, but can occur years after
- sx mild and nonspecific, and sx can be absent, but may have upper quadrant pain if severe rejection
- sometimes can have inc WBC +/- inc eos
- may be triggered by change in immunosuppressive meds or stuff that upregulates the immune system (such as infx)
- risk factors: inc donor age, younger recipient age, underlying autoimmune dz (PSC or AI)
- Labs: abrupt inc in liver enzymes above baseline, alk phos inc more than AST or ALT (which are also slightly inc)
Micro: portal inflam, bile duct injury, endothelialitis
-- need 2 of these 3 findings to fulfill Banff criteria
- endotheliitis can be absent, esp if late onset of acute cellular rejection
- portal inflam usually lymphs, but can have eos or plasma cells, and lymphs can appear "activated" (larger c irreg nuclei)
- bile duct injury includes inc lymphs and apoptosis, or can have reactive changes
- endothelialitis dx'd when portal veins or central veins have inc lymphs next to endothelial cells or in the lumen adherent to endothelial cells, causing endothelial cells to look reactive
IHC: not necessary not specific to make dx
DDx: acute rejection vs recurrent dz, must incorporate other lab tests
- recurrent hep C important to exclude, but can have significant overlap
- drug rxn usually c more eosinophilia
Lobular-Based Rejection
Predominantly lobular hepatitis, mostly in kiddos and adults that completely stop taking their meds
- Micro: lboules c mild to patchy mod lymph hepatitis c scattered apoptotic bodies and mild inc Kupffer cells
- portal tracts c mild lymph inflam, central vein not involved
- DDx: recurrent viral hep, EBV hep, drug rxn
Central Perivenulitis
- lobules c mild hepatocellular loss in zone 3 along c mild lymph inflam
- extravasated RBCs can be seen in this zone 3 region
- endothelium of central vein usually not c features of endothelialitis and is usually normal
- is usually accompanied by more typical acute cellular rejection changes in the portal tracts, tho can be an isolated finding
- can lead to central vein fibrosis and assoc c ductopenia
Chronic Hepatitis Pattern
Bx x chronic portal (zone I) inflam c minimal duct injury and no endothelialitis, ["piecemeal necrosis" not used anymore bc mechanism of cell death in interface hepatitis is apoptosis and not necrosis]
- aka idiopathic post-transplant hepatitis
- portal chronic inflam usually mild to mod inc lymphs, tho can have inc plasma cells;
- mild interface activity common, esp if mod portal chronic inflam
- cannot be confidently dx'd in pts transplanted for chronic hep C
- is probably a mix of dz's
Plasma-Cell Rich Rejection
If there is an inc in portal plasma cells along c otherwise normal findings of acute cellular rejection (duct injury +/- venulitis) is most consistent with acute cellular rejection
- inc plasma cells may have inc risk of acute plasma cell rejection
- inc plasma cells can also be caused by interferon-based tx, or autoimmune dz's (SLE) and can look like AI
- tx for rejection and AI are similar but not identical, and tx guided by the response to tx and not just histo findings (ab titers usually low or negative in plasma-cell rich rejection and are high in AI)
Antibody (Ab) -Mediated Rejection
Rare in liver; Graft injury caused by circulating abs to donor ag
- significance not well known in the liver, tho is important in kidney
- can only confidently dx when (1) donor-specific abs present, (2) requires clinical and lab-proven graft dysfunction, (3) active graft injury c histo features of ab-mediated graft rejection, and (4) C4d staining
Hyperacute rejection in liver, c preformed ABO abs, can happen after several hours or days (rather than minutes like other organs); rare
In ab-mediated rejection, have abs against lymphocytotoxic abs and anti-endothelial abs
- in first 2 weeks after transplant, in primary ab-mediated rejection, see early and significant graft dysfunction
- in secondary ab-mediated rejection, new (de novo) abs form after transplant, and can see acute cellular rejection with chronic rejection
Targets for abs can be determined in some cases, sometimes from glutathione-S-transferase T1 (GSTT1) or inherited genetic dz (bile salt export proteins), which the recipient ultimately recognizes as foreign
- actually considered part of autoimmune hepatitis
Micro: Hyperacute rejection (severe primary ab-mediated rejection)shows endothelial injury, microvascular thrombi, sinusoidal dilation, congestion bc endothelium is the target
- Acute ab-mediated rejection incompletely defined, but can basically show early changes (nonspecific, look like preservation injury [zone 3 hepatocyte ballooning, lobular spotty necrosis, cholestasis]; and later changes (mimicks biliary obstruction c portal tract edema, ductular prolif, portal neutrophilia)
IHC: C4d (immunofluorescence more sensitive [?}) in portal veins and or in sinusoids; best to see in a diffuse pattern (>50%)
- can also be seen in some allograft liver bx c no evidence of ab-mediated rejection; usually more focal
Px: eventually causes portal vein and hepatic artery thrombosis and ischemic necrosis of bile ducts and hepatic parenchyma
Chronic Rejection
Caused by chronic injury to bile ducts or liver endothelium
- causes chronic cholestasis in pt c no evidence of obstructive liver dz, drug rxn, or other cause of cholestasis
- imaging may show "pruning" (less small branches of intrahepatic bile tree)
- alk phos chronically elevated, maybe chronic AST or ALT inc
- pt more at risk if has had prior episodes of acute cellular rejection esp if refractive to tx
- usually occurs >60 days after transplant
- not possible to separate acute and chronic rejection on histologic exam only
Micro: need at least one of 3:
(1) >50% bile duct senescence,
(2) foam cell obliterative artiopathy
(3) >1/2 of bile ducts c bile duct loss
- bile duct changes are MC, foam cells rarely seen
- earliest change is bile duct senescence, which have smaller diamters than expected for the size of the portal tracts (remember that this happens normally in a certain % of bile ducts, so need > 1/2)
- may seen nuclear enlargement and uneven nuclear spacing
- med and large hepatic arteris have intimal thickening or foam cell obliterative arteriopathy in chronic rejection
IHC: CK may help identify bile ducts
DDx: must r/o obstructive biliary dz (such chronic biliary anastamosis strictures)
Biliary Obstruction
Impairment of bile flow caused by constriction of bile duct lumen that causes clinical or lab abnormalities
- clinical, imaging, and lab findings identical to those of non-transplanted livers c the same condition, c cholestatic pattern of injury c inc alk phos and inc bilirubin
Micro: similar to those of non-transplanted pts, c portal edema, bile duct prolif, portal neutrophilia in early stage
- later cases can have ductopenia and fibro-obliterative duct lesions
DDx: fibrosing cholestatic hep C, or marked hepatitis of any cause
- ab-mediated rejection has similar findings
- should correlate c imaging regardless
Hepatic Artery Insufficiency
Problems include anastomotic stenosis or thrombosis and occur usually in first 3 mo after transplant (median time 7 days)
-- frequency of hep artery thrombosis is 8% in kiddos and 3% in adults
- risk factors are CMV-seropositive donor to CMV seronegative recipient, prolonged operation time, retransplant, unusual arterial anatomy, transplant centers c low volumes
- early on, may see mild to mod inc AST or ALT, imaging may show stenosis or thrombosis
Micro: 2 categories: early vs late changes
- (1) early hetpatic artery thrombosis shows inc lobular hepatocyte spotty necrosis w/o sig inflam but inc cell cycling c inc mits, and inc apoptosis from ischemia
- (2) late changes in hepatic arterial thrombosis (weeks or months) range from coagulative necrosis to zone 3 hepatocyte dropout, bile duct injury c ductular rxn
DDx: recurrent hep C
- if mits prominent, favor arterial flow abnormalities
- preservation changes MC in first week after transplant
Misc. Vascular Problems
Small for Size Graft
Small liver allograft may not handle portal vein blood flow well, esp if graft <30% of expected liver vol or <0.8% of recipient's body weight
- may present c cholestasis, coagulopathy, ascites
Micro: early changes can have prominent portal veins and zone 1 sinusoidal dilation c bleeding into portal tract
- findings MC in larger portal tracts, may not be sampled in liver bx
- later on in dz, can have arterial vasospasm or thrombosis causing necrosis of large bile ducts in liver hilum which cause bile leaks, abscesses and parenchymal infarcts in liver hilum
- in less severe rxns may see nodular regen hyperplasia and duct rxn
Congestive Hepatopathy 2/2 Piggyback Graft
Piggybacking: Donor liver transplanted with intact recipient vena cava
- 1/10 of these pts get reactive ascites
- Micro: patchy zone 3 dilation and hepatocyte dropout, hepatic artery may be atrophied or inconspicuous
Viral Infection
MC is CMV which infects endothelial cells, bile ducts and hepatocytes
- Micro: may be mild and nonspecific c small foci of lobular lymph inflam to clusters of sinusoidal neuts (microabscesses, nonspecific)
May also get HSV and adenovirus
- both may cause azonal necrotic areas diffusely
- may have viral inclusions at edge of necrosis
Recurrent Hepatitis C
HCV recurs in almost all liver allografts after transplant
- dx made when liver injury assoc c Hep C infx, not when blood PCR has HCV RNA +
- HCV reinfects allograft in a few hours 2/2 circulating virons in blood
-- refractory time of weeks/months where liver enzymes steadily inc, which is usually not assoc c virus levels usually
Micro: vary depending on timing
- early changes are lobular patches of necrosis w/o inflam, which is assoc c first inc in AST and ALK (weeks or months after transplant)
- later changes shows less lobular apoptosis but mild lobular lymph inc, portal tracts c mild to mod chronic inflam
- same as in pts c chronic HCV
-- progression of fibrosis more rapid in non-transplanted pts
-- may score c Hepatitis Aggressiveness Score (HAS) for fibrosis progression
IHC: not useful
DDx: preservation injury, tho can be ruled out by timing of bx, and liver enzymes would be more stable
Alcoholic Liver Dz
- good indication for transplant
- same dx criteria as in nontransplant alcoholic liver dz
Non-alcoholic liver Dz
NAFLD commonly recurs in 5 years after liver transplant, and inc c time, maybe occurring in all livers
- assoc c metabolic syndrome, and steroid use as immunosuppressant
- may get fibrosis
- mild steatosis assoc c glucocorticoid tx for rejection control
Glycogen Hepatopathy
Hepatocytes swollen from glycogen accumulation, which can be caused by steroids, and also increases serum enzyme levels
- resolves c steroid tapering
Drug Rxn
Usually hard to recognize bc pt is taking lots of meds, and must r/o rejection and recurrent dz
- eosinophilia and portal lymph inflam may suggest
Graft Versus Host Dz (GVHD)
Seen after allogeneic BMT when engrafted donor's immune cells attack recipient liver
- shows clinically as gut, liver, skin or other injury to mucosa
- cholestatic patter c inc alk phos are MC
Micro: bile ducts injured and apoptotic
- inflam usually mild, without hepatitis
Engraftment syndrome - occurs in <100 hours after engraftment and is caused by release of cytokines by recently engrafted cells
- pt shows fever, rash, rapid weight gain
- should mimic veno-occlusive dz (sinusoidal obstructive syndrome)
Post-transplant Lymphoproliferative Disorder (PTLD)
usually EBV-driven in B cells, though 1/10 neg for EBV
- can significantly vary, but usually shows portal chronic inflam, sometimes c discreet B cell nodules
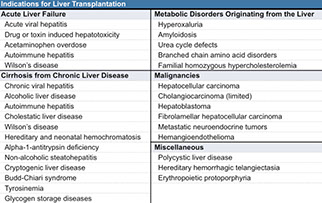
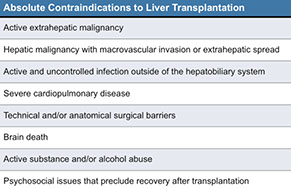
Severe preservation injury results in marked cholangiolar proliferation; acute cholangiolitis, hepatocellular swelling and hepatocanalicular and cholangiolar cholestasis. May persist for weeks to several months after transplantation. Similar changes can be induced by sepsis.

Acute Cellular Rejection


Chronic hepatitis c lymphs and plasma cells (arrow)

Inerface herpatitis (previously known as "piecemiel necrosis" is extension of inflam to periportal hepatocyte region. This would be a grade 2 interface activity (arrow)
Acute cellular rejection showing mixed portal infiltrates, easily identified eosinophils, lymphocytic bile duct injury, and endotheliitis / venulitis

Loss of small hepatic artery ba\ranches, chronic rejection

Chronic rejection, bile duct loss

From grand rounds presentation, Dr. Sarangarajan Ranganathan
Hepatic adenoma
MC in females 20-44 yo (the OCP years)
- inc androgen use also assoc c adenomas, also glycogen storage diseases type I and III
- the terrm "adenoma" may be used for any well-diff hepatic neoplasm
Sx: RUQ pain, but usually asymptomatic and found incidentally on imaging, which is solitary, well-circumscribed, no central scar
- may rupture and cause peritoneal hemorrhage
Risk factors: OCP use, anabolic roids, glycogen storage dz types I / III
Micro: resembles b9 hepatocytes, 1-2 cells thick cords
- clear cell change from glycogen; not encapsulated; no atypia, rare mits
- may be difficult to differentiate from background liver
- no portal triads
IHC: (+) ER/PR
- negative CD34 / factor VIII capillarization, negative glypican-3, glutamine synthase, HSP-70
- preserved reticulin framework (vs abnormally thick in HCC)
4 subtypes:
1) HNF-1a mutation - assoc c large droplet steatosis w/o atypia
- negative for all immunostains
2) B-catenin mutations - (+) atypia, and cholestasis, may transform to HCC
- (+) for B-catenin, Glutamine synthetase, Liver fatty acid binding protein
3) Interleukin 6ST mutation - aka inflammatory or telangiectatic adenoma
- strongly (+) for serum amyloid A, and C-reactive protein, also liver fatty acid binding protein
- pts c GNAS somatic mutations and McCune Albright syndrome may be predisposed to Inflammatory Adenomas
- should do B-catenin stain in these lesions to see if there is a B-catenin mutation ... may have inc chance of transforming to HCC??
4) Unclassified: No known mutation; no special features
Gene: HNF-1a at risk for MODY3 diabetes
DDx: Well-diff HCC (adenomas have no atypia / mits (low Ki67), no nucleoli and no reticulin loss)
- HCC can have IHC staining patterns that mimic adenoma subtype staining patterns
Tx: D/C OCP, serial imaging +/- AFP, +/- resection (if > 5 cm)
Px: 10% (?) malignant transformation (HCC), usually if >5 cm
Focal Nodular Hyperplasia (FNH)
2nd MCC benign liver tumor (after hemangioma)
Seen in young females, assoc c hyperplasia of hepatocytes caused by hyperperfusion due to hemangioma or vascular malformation (not really OCP use), 2/2 vascular shunting in general
- asymptomatic, no risk of hemoperitoneum
- in kiddos, usually preceded by chemotx
- may be seen in allografts
- does not impede flow of bile much, so only mild cholestasis, though some can have copper accumulation
- seen in lots of causes of vascular flow abnormalities, such as inflow dz (Abernethy syndrome), intraparenchymal dz (focal vascular shunts), chronic outflow dz (Budd-Chiari syndrome)
Gross: solitary, central scar
Micro: vascular lesion c arteries in fibrous stroma (lots of vessels in the scar) with fibrous bands separating benign hepatocytes; thickened plates of hyperplastic hepatocytes c eccentric muscular hypertrophy and prolif of bile ducts
- divides liver into nodules - focal cirrhosis
- cholestasis, bile ductules, copper (??)
- fibrous bands may resemble bridging fibrosis
-- ductular metaplasia - caused by atypical bile ductules growing in the bands
- central and peripheral regions have vessels c thick walls
- most cases >4 cm have a central scar
- subset c ballooning and Mallory hyaline, mimicking HCC (may use gluamine synthetase staining to help differentiate [has maplike / geographic staining in focal nodular hyperplasia, and is negative in HCC])
- background should not have cirrhosis (can mimick FNH)
IHC: (+) reticulin (hepatocyte plate of normal thickness [1-2 cells thick] vs thicker in hepatocellular ca, CK7 and 19 will highlight the bile duct proliferation
- neg: CD34, factor VIII, glypican-3, p53
Tx: observation (usually does not do anything) vs resection
DDx: inflammatory adenoma (telangiectatic adenoma)
- FNH-like region around an area with vascular shunting
Px: no malignant potential
Macroregenerative Nodule
B9 nodule in cirrhotic liver
- size makes it noticeable from background liver
- seen in ~1/7 explanted livers
- may also be seen in fulminant hepatitis
- can be positive for Glypican 3 (GPC3)
Dysplastic Nodule
Cirrhotic nodule which has some malignant characteristics, stands out from background cirrhosis; hard to define histologically but definitely does not meet criteria for HCC
Micro: may have pseudogland formation, mild atypia, few mits, intraparenchymal arterioles, complex architecture, nodule in a nodule appearance, usually c portal tracts present in the dysplastic nodules
• Clusters of dysplastic hepatocytes >1 cm in diameter
• Normal thickness trabeculae
• Portal structures in nodules
– Increased unpaired arteries
– Sinusoidal capillarization
IHC: will not have reticulin loss, negative AFP and B-Catenin
• No glypican, glutamine synthetase or HSP70
Vascular Malformation
Vascular mass lesion assoc c a vascular shunt, that cause sx in the first weeks after birth such as cardiac or resp failure (from hemodynamic compromise) and classically a bleeding diathesis 2/2 plt sequestration / consumption (Kasabach-Merritt syndrome)
- usually not bx'd and can have significant overlap c infantile hemangiomas
- have large cystic spaces filled c blood, organized thrombi and fibrous tissue
- IHC: GLUT-1 neg in vascular malformations but pos in infantile hemangiomas
Infantile Hemangioma
- previously infantile hemangioendothelioma, or diffuse hemagiomatosis (if multiple)
MC vascular tumor of liver in infants and toddles, b9 vascular neoplasm of pediatric liver; most in pts <6 mo age; 2F>1M
- ~1/2 c nonspecific sx (fail to thrive, GI probs), assoc c lots of other random congenital abnormalities
- tumors grow rapidly after birth then involute in childhood
- rarely cause death from heart of liver failure
Micro: well-demarcated, non-encapsulated, 1/2 are solitary lesions, entrapped bile ducts at periphery of tumor, 1/3 infiltrative at borders
- hepatocytes at margins may produce AFP
- the center of the tumor can look like a caavernous hemangioma
- the neoplastic endothelial cells are plump, but have no atypia
- can have high mits, but does not affect px
IHC: (+) CD31, CD34, GLUT-1
DDx: vascular malformations (GLUT-1 neg)
Tx: meds first, then excise if doesn't work
Px: can turn malignant, so should be evaluated for atypia, such as papillary tufts, solid areas, spindly kaposiform areas (although usually still are pretty b9 even if there is atypia)
Mesenchymal Hamartoma
b9 mass lesion of liver made of loose connective tissue usually c cystic change, c mixed in b9 bile ducts and someitmes small islands or cords of hepatocytes, most occur <3 yo and can be seen c prenatal US, usually single but may be multiple; unclear etiology
- usually present c abd mass, but can have a wide range of sx
- some can have inc AFP
Micro: usually made of loose CT c interspersed b9 bile ducts and sometimes small hepatocyte islands
- bile ducts are bland w/o atypia, no atypia also in stroma
- some can have a ductal plate malformation, or have dilated ducts c small cysts, and the larger cysts may not have an epithelial lining
- can see small islands of hepatocytes, and absent or mild inflam
- commonly see EMH
IHC: ductular epithelium is CK7 and CK19 (+)
- loose CT (+) vimentin and smooth muscle actin
(+) glypican 3 in hepatocytes
Px: can rarely undergo malig transformation to embryonal sarcoma
Hemangioma
MC benign liver tumor, seen in 1/20 livers, MC in young adult women, can enlarge c preg or E therapy, dx usually made on inmaging and no bx is needed; 9/10 are solitary tumors
- only rarely become significant if rupture 2/2 trauma or if enlarge and breach their capsule
- may cause pain if >4 cm
- if a cavernous hemangioma is >8 cm, called a giant cavernous hemangioma, and has similar micro findings as regular cavernous hemangioma except possibly ill-defined borders (called hemangiomatosis or hemangioma-like vessels)
Gross: solitary, red-brown, well-circumscribed spongy lesion
Micro: most are cavernous hemangiomas c well-circumscribed unencapsulated aggregate of large-caliber, thin-walled BVs which are supported by network of fibrous stroma and lined by thin layer of endothelial cells w/o atypia or mits
- Blood filed vascular spaces of various size lined by a single epithelial layer and can have fibrin thombi in them and become infarcted, and might show lots of macrophages
Capillary hemangiomas (aka lobular hemangiomas) have slight female predominance, and shows small thin-walled vessels in lobular arrangement and may have a more solid appearance 2/2 very small vascular lumens
- tumor cells are plump w/o atypia or mits, and can have EMH
- IHC: (+) CD34, ERG (in endothelial cells)
Dx: Do NOT bx bc can cause fatal hemorrhage, can FU with MRI
Px: excellent px, no malig potential, some may entirely regress c fibrosis over time
- giant hemangiomas assoc c Kasabach-Merritt syndrome, rare complication of vascular tumor and consumptive coagulopathy
Epithelioid Hemangioendothelioma (EHE)
Low-grade malignant vascular tumors made of epithelioid and dendritic tumor cells in a myxoid or hyaline matrix
- can also be in lung (IV-BAT) and ST
- F>M, possible a/w OCP, but not cirrhosis, avg 45 yo
- often multiple, in both lobes, ~1/2 are incidental findings
- low-grade endothelial tumor that may metastasize
Micro: infiltrates sinusoids and veins, tuft-like intravascular prolif and as fibrothrombotic occlusions (extensive fibrosis occluding hepatic and portal vein branches)
- plump epithelioid cells or signet-ring type cells c acidophilic cytoplasm, vacuoles and RBC (signet-ring cells are mucicarmine neg)
- abundant myxoid or sclerotic stroma
- tumor cells are usually denser at the periphery, and can become sclerotic c calcifications
- may show areas of necrosis and hemorrhage
- mits rare or absent
- liver capsule is involved in ~1/2 of cases
Gene: t(1;3)(p36;q25)
- this is a dz-defining translocation and can be useful in a difficult case
IHC: (+) CD34, FVIII, CD34, CD31, FXIIIa, ERG are all positive in almost all cases; CD10+ in most cases (can cause confusion on small bx's)
DDx: are often misdiagnosed as cholangiocarcinoma (probably from signet ring cells and lots of extracellular matrix)
- angiosarcoma and other carcinomas (HCC) can also look similar
Px: low-grade malignancy (~25% met, ~20% 5-yr survival)
- inc cellularity is the strongest predictor for aggressive behavior (although all are malignant and can be aggressive)
Angiosarcoma
High-grade malig vascular tumor c several growth patterns
- rare and can mimic other tumors, can be primary or metastatic
- risk factors: arsenic, androgen tx, Thorotrast, vinyl chloride
- no cause found in 7/10 cases
- usually occurs in older men
Micro: can be single or multiple
- background liver c chronic inflam or fatty change, can have little fibrosis or cirrhosis
- grow as vascular tumor c irregular BVs or solid epithelioid tumors that mimic carcinoma or spindle cell tumors that look like other sarcomas
- has small slit-like vascular spaces c RBCs, and usually has lots of atypia and mits
- solid parts may undergo necrosis and cavitation, causing a rim of malig cells around a cavity c blood, fibrin and necrosis
- may grow along sinusoids, leaving the normal liver architecture somewhat intact (high Ki67 may help prove it is angiosarcoma)
IHC: (+) FVIII (up to 9/10), CD34 (3/4), CD31 (1/3), ERG
- up to 1/2 have aberrant AE1/3 or CAM5.2 (+)
Px: dismal (<6 mo)
Angiomyolipoma
B9 mesenchymal tumor c myoid cells mixed c fat and big irreg vessels
- most are sporadic and solitary (9/10) and not part of tuberous sclerosis (though 1/10 are assoc c TS), avg age 50 yo, F>>M
- can rarely be malig (if has vascular invasion or aggressive behavior such as mets)
Micro: made of fat, sm muscle, or myoid differentiation (epithelioid or spindly cells) c thick-walled BVs
- background liver usually w/o dz or fibrosis
- some tumors are made of mostly fat, and can look like a lipoma or liposarcoma
- in tumors c epithelioid predominance, may resemble HCC or a clear cell tumor
- may have EMH, hemorrhage, necrosis, cholesterol clefts, giant cell change, inflam (looking like an inflammatory pseudotumor), or atypia (does not indicate malignancy)
IHC: (+) HMB45 (can be patchy, esp in fatty areas), MelanA (in 9/10), smooth muscle actin (esp in spindle cell areas), CD117 (almost all cases), CD68
- neg CK and Hep-Par1
DDx: GIST (also CD117 +)
- fibrolamellar ca (has CK7 + and HMB45 neg)
Solitary Fibrous Tumor (SFT)
B9 spindle cell tumor of unknown etiology, usually in middle-aged women, but is variable; may cause hypoglycemia from production of insulin-like growth factor 2
- can be multifocal, despite name
- are usually intraparenchymal, only a few are assoc c the capsule
Micro: has hypo- and hypercellular areas (similar to Schwannoma) of spindly cells, but also a thick ropey collagen
- low cellularity c tumor cells in fibrous stroma, in a patternless pattern
- some may become sclerosed
- sometimes can entrap bile ducts and cause secondary biliary cysts
- proliferating bile ducts at periphery of lesion can show pancreatic or hepatic metaplasia
IHC: (+) Bcl2 and CD34
- neg: S100, desmin, CD117, smooth muscle actin, CKs
Px: May transform into high-grade fibrosarcoma (assoc c inc mits, necrosis and atypia)
Segmental Atrophy of the Liver
B9 pseudotumor assoc c parenchymal loss and replacement by elastosis and fibrosis
- MC in subcapsular, F>M, pts present c RUQ pain, can be up to 10 cm
Micro: findings evolve over time, so depends on age of lesion
- early lesions c parenchymal collapse c marked bile ductular prolif and mixed inflam
- over time get less inflam and ductular prolif and inc amts of elastosis in areas of parenchymal loss, which is assoc c retention-type biliary cysts (which can rupture and cause inflam response)
- nodular elastosis is the stage where elastosis is the predominant finding, and has islands of normal-looking hepatocytes, and reticulin stains highlight the inc reticulin fibers
-- the elastosis usually involves the liver capsule
-- high power shows inc spindle cells w/o atypia or mits which are vimentin + and highlight the dendritic extensions of the tumor cells
- may have small calcs, and thrombosed and fibrotic vessels are common (suggesting a vascular etiology)
Small areas of elastosis may be incidentally found on liver bx, these have no known significance
Inflammatory Pseudotumor
- aka Inflammatory myofibroblastic tumor
B9 reactive pseudotumor made of varying degrees of fibrosis and plasma-cell rich chronic inflam
Presents c fever, abd pain in all ages
- 2M>1F, ~50 yo, have been linked to all sorts of stuff (tumors, infx, chronic liver dz, autoimmune dz)
- may have inc CA19-9
- can regress spontaneously or c abx tx
- usually solitary lesions (2/3)
Gross: variegated c hemorrhage and necrosis
Micro: single lesions usually bigger than multiple lesions, and are more common in non-cirrhotic livers
- made of mix of fibroblasts and inflam cells c varying collagen (which can be dense and whorled)
- spindle cells should have no atypia, and mits are rare
- inflam has lots of plasma cells and T cells, B cells limited to germinal centers of lymph aggs
- occlusive phlebitis in med and large portal veins common in resection specimens
- may have scary giant follicular dendritic cells (assoc c EBV)
IHC: spindle cells have (+) vimentin and smooth muscle actin, patchy CK
- neg ALK (in 100%)
DDx: lots of stuff can mimic inflammatory pseudotumor (lymphoma, angiomyolipomas, liposarcomas, carcinomas, cholangitis)
- should do workup on pt to exclude these dz's if dxing inflammatory pseudotumor
Embryonal Sarcoma
- aka undifferentiated embryonal sarcoma, hepatic undifferentiated sarcoma
Undifferentiated sarcoma MC in peds pts, M=F, avg ~10 yo
- can be seen in adults, unknown etiology, may arise from mesenchymal hamartomas
Micro: undifferentiated spindle cells c diffuse atypia; cells are med- to large-sized and can have giant cell transformation
- cellularity can vary, but stroma is usually loose and myxoid
- may see hyaline globules in tumor cells, which are PAS-(+)
- may see remnants of a mesenchymal hamartoma, esp in young pts
IHC: diffuse (+) for CKs such as AE1/3 and CAM5.2 (which can have perinuclear dot-like +), large multinucleated cells can be glypican-3 +
- neg: myoglobin (can be focally +), smooth muscle myosin, h-caldesmon, CD34, ALK1, S100, GFAP
DDx: biliary tract rhabdomyosarcoma (lack diffuse anaplasia and hyaline globules and are MyoD1 +)
- metastatic sarcomas in adults must be r/o'd
Hepatoblastoma
Malig epithelial tumor sometimes c sarcomatous component, usually presents in 1st 3 years of life c abd mass or swelling
- assoc c FAP, Wilms tumor and Beckwith-Wiedmann
- other risk factors are prematurity and low birth weight (esp <1500 g), 2M>1F, and sometimes occur with other genetic syndromes
- MCC malignant liver tumor in 1st 5 yrs of life
- present c large abdomen, weight loss, anorexia, rarely have paraneoplastic syndrome
Micro: Mixed epithelial and mesenchymal ("light-dark" pattern of thickened cords of cancerous hepatocytes), extramedullary hematopoiesis
- subclassified into epithelial (further broken down into fetal and embryonal types) and mixed epithelial-mesenchymal types
- squamoid differentiation can be seen after tx
Genes: mutations in proteins that code for Wnt signalling pathway, esp B-catenin (can show abnormal nuclear or cytoplasmic + by IHC), also AXIN1, AXIN2 or APC genes can be mutated
- MSI in 4/5 cases, TP53 in 1/4
Dx: nonspecifically deranged liver enzymes, inc B-hCG, inc aFP in 90% (usually markedly inc)
- US good for screening, CT good to assess operability
DDx: metastatic small round blue cell tumors (Wilms tumor and neuroblastoma)
- Transitional Liver Cell Carcinoma (subtype of HCC in older kiddos usually mistaken for hepatoblastoma, thought to be somewhere in transition bwt HCC and hepatoblastoma)
Tx: complete resection (chemo/rads also works)
- tumor is usually shrunk prior to excision
Px: mostly depends on if it is completely resected
- Worse if has low levels of AFP, or if it has rhabdoid features
- angiolymphatic invasion may portend poor px
Fibrolamellar Hepatocellular Carcinoma (FLC)
Younger age (10-35 yo), M=F, no hx of cirrhosis or other risk factors; L>R, presents as large solitary mass (>10 cm)
- ~1/20 cases of HCC
Labs: usually normal AFP, inc transcobalamin 1 and 2, inc vit B12 binding capacity, inc serum fibrinogen and neurotensin
- inc des-gamma-carboxy prothrombin (DCP)
- activation of protein kinase A, usually through a DNAJB1-PRKACA fusion transcript
Gross: central scar (in 7/10 cases)
Micro: no sinusoids / nests; parallel lamellar fibrosis, tumor cells larger than HCC, oncocytic eosinophilic cytoplasm, mitochondia, giant nuclei, prominent nucleoli, 50% c cytoplasmic pale bodies inclusions (stain for fibrinogen)
- can have small eosinophilic inclusions (hyaline bodies) in half of cases
-- hyaline bodies and pale bodies are NOT specific for FLC
- can have epithelioid granulomas
- may have copper deposition from cholestasis
- some can show glandular structures c mucin, which are cystic to ovoid
- mets to LNs usually have the same structure as the primary tumor
- vascular invasion frequently seen (25% grossly, 50% microscopically)
IHC (+): CK7, HepPar1 (strongly +), rarely glypican 3 (+), CD68 (strong + from tumor lysosomes) - CD68 highly sens (96%) and spec (80%) for FLC, and double negative staining of CK7 and CD68 excludes FLC [1]
- neg: aFP
Genes: activation of protein kinase A, usually 2/2 DNAJB1-PRKACA fusion transcript
DDx: cholangioca (from intratumoral fibrosis and possible mucicarcmine production)
- scirrhous HCC (CD68 neg and usually assoc c chronic liver dz, not seen in FLC)
Px: better than HCC (43 mo vs 6.5 mo), depends if it can be resected
Intermediate Cell Carcinoma
Morphologically defined, immunophenotypically confirmed
- biphenotypic
Micro: monomorphic tumor cells intermediate bwt hepatocytes and cholangiocytes
- cords, nests, trabecular
IHC: CK7 (
Heptaic adenoma

Hepatic adenoma, well-circumscribed,

FNH




1 - 3
<
>
Macroregenerative nodule

Proliferating infantile hemangioma

Mesenchymal hamartoma
Hemangioma


Epithelioid Hemangioendothelioma


EHE
Angiosarcoma

Angiomyolipoma


SFT

Segmental atrophy of liver


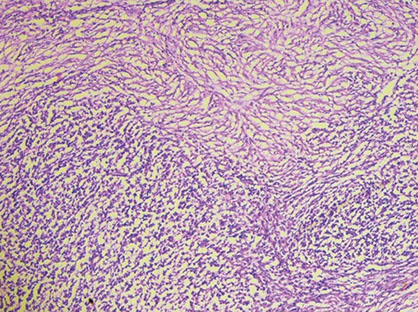
Inflammatory pseudotumor

Inflammatory myofibroblastic tumor



Embryonal sarcoma - Intracytoplasmic and extracellular eosinophilic globules, are PAS-D +
Embryonal (undifferentiated) sarcoma


Hepatoblastoma
Hepatoblastoma
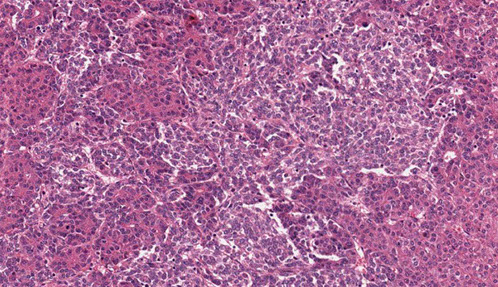
Fibrolamellar Hepatocellular Carcinoma



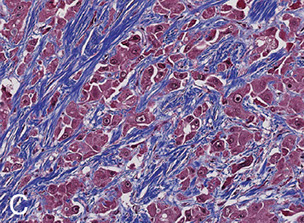
Trichrome stain highlights lamellar fibrosis (C)
(D) Large polygonal cells c prominent nucleoli and abundant eosinophilic cytoplasm. Pale bodies (red arrow) and hyaline bodies (black arrow) seen [1]

CD68 in FLC showing weak granular cytoplasmic staining [1]

Intermediate Cell Carcinoma


Hepatocellular carcinoma
MC primary malig tumor of liver in adults; though relatively uncommon overall (mets are MC in liver) - at least in US [some pops are much more inclined, like Asians and other places c lots of HBV]
- mean age: 61 yrs; M:F = 4:1; 80% c hx of cirrhosis
Risk factors: Hep B / C, Wilson's dz, hemochromatosis, a1-antitrypsin def., EtOH cirrhosis, polycythemia (inc EPO), hypoglycemia (inc insulin-like growth factor), hypercalcemia (inc PTH-rP)
Micro: may be uni- / multifocal or diffuse
- likes to invade vascular channels
- patterns: trabecular, pseudoglandular, compact/solid, fibrolamellar (discussed above), cirrhosis, well/poorly differentiated
- more poorly diff tumors have increasingly more basophilic cytoplasm and worse nuclear atypia
Dx: Bx
- inc AFP (also in gonadal tumors)
IHC: stains used for 2 purposes (1) to tell a well-differentiated tumor from b9 liver, (2) to tell a poorly diff tumor from mets
(+) Glypican-3 (GPC-3, can be neg in well-diff HCC) and CD34 diffuse staining (vascular pattern and sinusoid-like spaces), factor VIII; Arginase-1 (Arg-1) and hepatocyte parrafin-1 (HepPar-1, pretty spec) to distinguish from mets, (+) CD8/18, (+) pCEA [apical / canalicular pattern], CD10/ villin (canalicular), CAM5.2, TTF-1 (cytoplasmic)
- reticulin stain loss
- AFP (1/2+, not specific, but generally negative in benign lesions of liver)
- negative CK7/20 (usually), AE1/AE3 (up to 1/5+), mCEA, p53 (var)
Arginase-1 sensitive and specific for normal hepatocytes and malignant hepatocytes (HCC)
- can also be positive in hepatoid adenocarcinoma
- may be useful in distinguishing HCC from cholangiocarcinoma
Tx: resect if localized; early transplantation
Px: aggressive and life-threatening (avg survival 6.5 mo)
- hematogenous metastasis
- Px depends on tumor resectability mostly, but also on pt hx and some path characteristics (tumor size, lymph-vasc invasion)
Biphenotypic HCC
- aka combined (or mixed) HCC and cholangiocarcinoma; previously "collision tumors"
Single tumors c 2 morphologies: one similar to normal HCC and the other like cholangiocarcinoma
- seen in ~1/50 HCC
IHC: (+) HepPar1 and Arginase-1 in HCC part, which is negative in cholangiocarcinoma part (which is + for biliary-type keratins)
- regular HCC may aberrantly be + for biliary-type keratins, should not call it a biphenotypic HCC unless there are 2 different morphologies
DDx: pseudoglands in typicall HCC can look like cholangioca
- robust ductular rxn can be seen at interface of a normal HCC and b9 liver parenchyma which should not be overinterpreted as biphenotypic HCC
Genes: combined HCC/CCA clusters more with cholangiocytes, while classic HCC clusters more with hepatocytes
Px: better than cholangioca, worse than HCC
Cirrhotomimetic HCC
- aka diffuse HCC, or cirrhosis-like HCC
Rare; Grows as small nodules that look a lot like cirrhosis
- can have a huge tumor burden that may look like cirrhosis on imaging and on gross exam
- not really known why it grows like this
- may have hematogenous spread
Micro: small blue tumor cells in small nodules spread throughout the liver
- may appear as cirrhotic nodules "transforming" into carcinoma
- usually mod to poorly-differentiated
Clear Cell HCC
>1/2 of tumor cells c clear cell changes
- may be up to 1/20 HCC cases
- usually well- to mod-diff,
- IHC is similar to that of normal HCC
- may be a different entity than normal HCC if there is 100% clear cell changes
- clear cell HCC with cytoplasmic glycogen may be different from HCC with cytoplasmic lipid accumulation
DDx: clear cell ca from other organs (kidney)
Px: may be better than conventional HCC
Granulocyte Colony-Stimulating Factor Producing HCC
Rare (<1/100); Has lots of neuts in the sinusoids
- usually in older pts, has a poor px
- presents c a very high WBC count, fever, inc CRP
- usually mod- to poorly-differentiated, possibly c areas of sarcomatoid changes
- areas of normal HCC that have been tx'd (c chemo or embolization) can have severe neut rxn
IHC: usually weak or patchy staining c normal HCC markers (but should be + somewhere to prove that it is from liver)
DDx: mets
Hepatic Stem Cell Ca / Hepatic Progenitor Cell Ca
Rare (<1/100); has 2 cell populations: 1 is usual HCC, other c primitive undifferentiated features
- Micro: has small undifferentiated cells mixed c more typical HCC cells
- these atypical cells usually stain differently by IHC (CK7/19 +)
DDx: Biphenotypic HCC (combined HCC and cholangioca)
Lymphocyte-Rich HCC
Rare (<1/100); Lots of lymphs inside tumor, usually dx'd if there are more lymphs than tumor cells diffusely
- the lymphs are reactive T cells by IHC, with B cells seen in germinal centers
- may be called lymphoepithelioma-like HCC (though may reserve this term for if there are syncytial sheets of epithelial cells)
DDx: lymphoma, though should be able to differentiate morphologically
- lymphoepithelioma-like cholangiocarcinoma (usually EBV-+ and does not stain with liver markers)
Px: better than conventional HCC
Sarcomatoid HCC
Spindle cell ca c focal hepatic differentiation, can be difficult on needle bx
IHC: (+) vimentin in spindle cells, which should be be at least focally positive for a hepatic marker
Scirrhous HCC
Rare; is HCC with ~1/2 fibrous area, and can happen in cirrhotic or non-cirrhotic livers, actually MC in non-cirrhotic livers
- usually subcapsular, though they do not have a capsule themselves
- can have fatty or clear cell change, and can have hepatocyte inclusions (as red or clear inclusions)
Micro: cancer cells grown in cords or trabecula which look atrophic
- fibrosis can have different patterns, from growing along sinusoids, to a more lamellar pattern, to a more clumpy pattern
DDx: cholangiocarcinoma or mets
Steatohepatitic HCC
HCC c features of steatohepatitis
- can be in a typical HCC that gains lots of fat in a pt c metabolic syndrome and also has steatohepatitis (9/10); or can occur as a distinct entity without inc fat but c distinct mutations without relation to metabolic syndrome (1/10)
Micro: see features of steatohepatitis such as fat, inflam, balloon cells, Mallory hyaline, pericellular fibrosis
Giant Cell Tumor of the Liver
Very rare; prolif of undifferentiated cells mixed c histiocytes forming giant cells; can have lots of hemosiderin deposits
- usually assoc c BilIN-3 lesions, suggesting relation to biliary tract dz
IHC: undifferentiated part of the tumor is CK-neg, not really sure where this tumor comes from


Biphenotypic HCC

Clear cell HCC
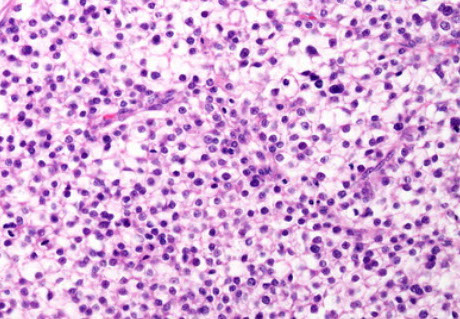

AJCC 8th edition staging for HCC (including fibrolamellar variant)

Gallbladder
Embryology
Biliary tree derived from endoderm, and epithelium of intrahepatic bile ducts comes from primitive hepatic cords
- formed by the 12th wk c secretion (excretion) of bile into duodenum by 16th wk
Anatomy
Normal max dimensions: 10 cm (length) x 3-4 cm wide, 1-2 mm wall thickness
- parts: fundus, body, infundibulum, neck
-- Hartmann's pouch: outpouching in infundibulum that may be caused by chronic inflammation
Histology
4 layers: 1) Mucosa, 2) SM 3) Perimuscular subserosal CT, 4) Serosa
- No muscularis mucosae nor submucosa
1) Mucosa: nonstratified columnar epithelium with oval, basal nuclei, absent nucleoli
- interspersed thin cells w/ magenta cytoplasm, dubbed "pencil cells"
- only neck has cuboidal, tubuloalveolar mucous glands
-- glands present in rest of g-bladda if chron. inflam
--- chron cholecystitis/ cholelithiasis causes metaplasia:
-"gastric" metaplasia: foveolar-type epithelium/ antral-type glands
-"intestinal" metaplasia (Paneth cells, absorptive cells [have brush border], endocrine cells, or goblet cells),
-squamous metaplasia
---- may differentiate normal g-bladda mucin from stomach/ intestinal mucin w/ IHCs
- arranged in uneven/branching folds into lumen having lamina propria core
-- lamina propria: loose CT, elastic fibers, VANs, lymphos
2) SM: 3 layers, like stomach, but not well-arranged
- ganglion cells found here (also in layers (1) and (3))
- may have Cajal cells (thus may get g-bladda GISTs)
3) Perimuscular subserosa: similar to lamina propria
- rarely, get a lymph node here
- also rare: paraganglia around vessels
- Rokitansky-Aschoff sinuses: invagination of epithelium that may rarely reach this level ([3])
-- more common w/ chron inflame, tho in normal g-bladda too (40%)
- Luschka's ducts: groups of microscopic ducts in this level ([3])
-- more common close to liver (may come from liver [??!])
4) serosa ???
Canals of Hering: are CK19 (+)

Cystic Duct
Surface cells identical to those of g-bladda
- pts w/ pancreatobiliary dz may have enterochromaffin cells w/ serotonin or somatostatin - (+)
- spiral valve of Heister: SM at cystic duct-gbladda junction that may prevent overdistention
Biliary Atresia
Caused by inflammation or toxic destruction of extrahepatic biliary tree, in the first weeks of life, presenting c jaundice, pale poop, dark pee, pruritis, failure to thrive (poor weight gain, excessive feeding)
- can be subdivided into 3 types (cannot distinguish from liver bx):
--Type 1 (1/20): atresia in common bile duct (CBD), left and right hepatic ducts and common hepatic ducts patent, causing dilation of the patent ducts, bile still in the gallbladder
--Type 2 (1/50 cases): common hepatic duct atresia, with dilation of the left and right hepatic ducts, no bile in gallbladder
-- Type 3 (>9/10): atresia of right and left hepatic ducts around the liver at porta hepatis
Labs: inc conjugated hyperbilirubinemia, inc GGT, inc alk-phos, inc cholesterol and normal triglyceride levels
Biliary atresia-splenic malformation syndrome: polysplenia, asplenia, or double spleen
- some can have cardiovascular malformations (situs inversus, preduodenal portal vein, absent vena cava), intestinal malrotation, pancreatic anomalies
Imaging: hepatomegaly c no dilatation of common bile duct, gallbladder can be absent, contracted or normal
- HIDA scan shows normal hepatic uptake, but poor intestinal excretion, though decreased secretion is not specific and can be due to other pediatric cholestatic liver diseases
Micro: histology depends on when the bx taken in relation to disease, may be normal early on, later can see bile ductular proliferation, some portal fibrosis
- hepatic artery usually prominent 2/2 thickened muscular wall
- up to 1/2 have proliferating ductules that have circular structure resembling early ducts in liver organogenesis, suggesting ductular plate malformation
- hepatic lobules c cholestasis, and little or no inflam changes
- giant cell transformation can be seen in hepatocytes
- EMH common
Hilar plate taken during Kasai procedure shows small-sized biliary ducts, sometimes peribiliary glands, lymph inflam, background of fibrosis and connective tissue
IHC: proliferating bile ducts are CD56 positive (usually not positive in other diseases)
Tx: Kasai procedure (hepatoportoenterostomy) - loop of duodenum attached to hilum of liver after removal of biliary ducts
- Kasai procedure is not curative, but used as a bridge for liver transplant; best outcomes seen in pts <60 days old, and may benefit from using corticosteroids, antibiotics, bile flow stimulators
DDx: type 1 biliary atresia and choledochal cysts can look similar on imaging studies
- Neonatal Giant Cell Hepatitis (NGCH) - both dz's can have giant cell transformation, portal fibrosis, ductular proliferation
Px: hilar plates c large-sized bile ducts (suggesting not as much atresia) tend to do better
Neonatal Giant Cell Hepatitis (NGCH)
Causes cholestasis in infants, bx shows giant cell transformation of hepatocytes, without evidence of biliary atresia or paucity of intrahepatic bile ducts
- clinically have inc conjugated bilirubin
- normal biliary trees on imaging
- MCC is idiopathic (1/2), then hypopituitarism, biliary atresia (1/10), Alagille syndrome, bile salt deficiencies (such as BSEP), neonatal hemochromatosis, cystic fibrosis, a1-AT def, type 2 Gaucher dz, CMV, echovirus, autoimmune hepatitis, SCID
Micro: syncytial giant cell transformation of hepatocytes (usually ~1/2 hepatocytes affected, usually zone 3 distribution or azonal)
- usually mild inflam, mild lobular cholestasis
- lobular EMH in ~3/4, sometimes mild fibrosis
Paucity of Intrahepatic Bile Ducts
Pathologic dx, but not really a specific entity, and is a pattern of injury with many different causes (similar to NGCH)
- may be 2/2 infx (CMV, HBV, syphilis), metabolic defects (BSEP, a1-AT def, CF, hypopituitary dz), genetic anomalies (tri 21/11/18)
- clinically c jaundice, conjugated hyperbilirubinemia, acholic stools
- HIDA can show failure to secrete into intestines
Micro: Bile ducts hypoplastic or absent in portal tracts, which can also have all kinds of inflam, and cholestasis, mild giant cells
- can have mild bile duct prolif
- fibrosis is uncommon
IHC: CK can help highlight loss of ducts
- CD10 shows loss of lobular bile canaliculi in older kiddos
DDx: histologic overlap bwt many dz's
Familial Cholestasis
AR, usually c family hx of liver dz
- historically classified by phenotype, but now by genetics
- all can cause intrahepatic cholestasis of pregnancy in heterozygotes
- all cases of bile salt deficiencies have inc serum conjugated bilirubin, and usually mildly inc AST and ALT
Micro: significant overlap bwt dz's
ATP8B1 deficiency
Relatively more episodic patterns of injury (formerly benign recurrent intrahepatic cholestasis or BRIC1, Byler dz, Greenland familial cholestasis), missense mutations generally do better (have residual protein activity) vs nonsense/frameshift mutations
- clinically c diarrhea, pancreatic dz and hearing loss
- has bland lobular cholestasis
- EM shows course granular bile (Byler bile)
ABCB11 (BSEP) deficiency
Relative more persistent injury patterns (aka progressive familial intrahepatic cholestasis or PFIC1 or PFIC2)
ABCB11 codes for bile salt export pump (BSEP) protein, with mutations causing impaired canalicular transport of bile salts
- typically c inc serum AFP
- has NGCH pattern
- significant risk of HCC and cholangiocarcinoma
- EM shows amorphous or filamentous bile
- IHC c dec or loss of BSEP
ABCB4 (MDR3) deficiency
Typically have inc serum GGT levels, usually present at older ages (~3 yo)
- MDR3 mutations caused impaired canalicular translocation of phosphatidylcholine
- resembles biliary obstruction, c dominant bile ductular reaction
- MDR3 loss by IHC
Cholecystitis
Gallbladder inflam usually due to gallstones by sometimes from ischemia or infx (E Coli, Klebsiella, CMV)
Sx: (+) Murphy's sign; should also check for peritoneal signs
- inc alk phos c bile duct involvement (ascending cholangitis)
Dx: US, possible HIDA
Tx: Cholecystectomy (usually delayed until after 24-48 hrs of medical tx)
- must open pt up if perforated (laparoscopic otherwise)
- ERCP for unstable pts c choledocholithiasis (stones in bile duct); bile acids / lithotripsy c mild sx
Cholelithiasis (gallstones)
Form when solubilizing bile acids and lecithin are overwhelmed by inc cholesterol, bilirubin, or gallbladder stasis (bile supersaturated c cholesterol)
Risk factors: Female, Fat, Fertile, Forty
Charcots triad: Jaundice, Fever, RUQ pain
- Reynolds pentad = Charcot's triad + HTN and altered mental status
(+) Murphy's sign
Types of gallstones: Cholesterol, Pigmented, mixed
1) Cholesterol stones (70%): radiolucent c 20% opacity due to calcifications
- assoc c obesity, Crohn's, CF, inc age, clofibrate, E, multiparity, rapid weight loss, Native American ancestry
2) Pigmented stones (20%): Radiopaque
- assoc c chronic hemolysis, EtOH cirrhosis, inc age, biliary infx
- black pigmented stones = inc bilirubin from extravascular hemolysis
- brown pigmented = sign of bile duct infx
Hepatic abscess
fever, RUQ pain, weight loss c nausea and vomiting
- pyogenic liver abscess: gets to liver by portal vein or biliary obstruction; caused by E coli, Klebsiella, or anaerobes
Tx: drainage and IV abx (broad spectrum)
Amoebic liver abscess
- caused by Entamoeba Histolytica (also in portal v.)
Tx: IV metronidazole and drain if necessary
Clonorchis sinensis and Opisthorchis viverrini
Ovoid egg that's moderately thick, bile-stained shell, prominent operculum c "shoulders"
- small knob at abopercular end (opposite operculum), egg contains a miracidium
Causes biliary fibrosis, recurrent suppurative cholangitis, cholangiocarcinoma
Clonorchis sinensis and Opisthorchis viverrini


Reye's syndrome
Rare but lethal (in 25%) childhood hepatoencephalitis assoc c VZV/ influenz B infx tx'd c salicylates (ASA)
- kids c inherited fatty acid oxidation defects at inc risk
Findings: mitochondrial abnormalities, microvesicular fatty liver, hypoglycemia, coma
Labs: inc ammonia, inc transaminases, prolonged prothrombin time
Mech: ASA metabolites dec B-oxidation by reversible inhibition of mitochondrial enzymes
*** Lesson: ASA Not recommended for children (use acetaminophen instead [with caution])
- exception is in kids c Kawasaki dz, consider giving VZV vaccine
Budd-Chiari syndrome
occlusion of IVC or all 3 hepatic veins c centrilobular congestion + necrosis leading to congestive liver dz (hepatomegaly, ascites, abd pain, and eventual liver failure)
- although can see varices and signs of portal HTN, there is no JVD (vs rt-sided heart failure)
- Assoc c polycythemia vera, pregnancy and HCC
Micro: intimal fibrosis, sinusoidal dilation,
Veno-occlusive disease (Sinusoidal Obstruction Syndrome)
Pts exposed to radiation or hepatotoxins (esp in pts preparing to get BM transplantation, but also caused by some alkaloids) getting obstructive lesions which are non-thrombotic in the small hepatic veins
- may be 2/2 sloughing from endothelial necrosis into veins
- sx: rapid weight gain, ascites, liver failure
Histo: bleeding into liver parenchyma early on in dz
- after several weeks see sinusoidal congestion and loss of zone 3 hepatocytes c sinusoidal and intimal fibrosis
Acute Biliary Obstruction
Pt presents c biliary colic that can radiate to back, sometimes assoc c eating fatty food
- pts typically have elevated Alk phos, GGY and bilirubin
- bx rarely done
Micro: bile ductular prolif, portal inflam c mixed neuts and lymphs, and usually portal edema
- this combo of findings is called "bile ductular reaction"
- neuts in a dilated lumen suggest additional finding of ascending cholangitis
- high-grade obstruction can sometimes cause bile infarcts
- special stains usually not needed, though ductules in cirrhotic livers c ductular rxn are C56, EMA and CD10 + whereas in acute obstruction they are CD56/CD10 neg and EMA +
Ddx: ductular rxn can be seen in several dz's, esp marked hepatitis or vascular outflow dz
Chronic Biliary Obstruction
Presents c cholestasis, such as inc serum bilirubin and alk phos
- may be vaused by stones, tumors, parasites, or pancreatic tumors that compress the bile ducts
Micro: depends on extent and duration of dz
- early or mild cases have patchy bile ductular prolif c mild mixed portal inflam (which is less than in acute obstruction)
- ductular rxn can dec c time, and the portal tracts may become ductopenic
- onion skinning fibrosis, bile duct duplication, and fibro-obliterative lesions more assoc c chronic dz
- zone 1 hepatocytes can show cholate stasis c mild hepatocyte swelling. sometimes mallory hyaline, and patchy copper accumulation; can be recognized on low power with the halo sign
may show pancreatic acinar metaplasia in peribiliary glands in bigger portal tracts
DDx: mild ductular rxn common in most cirrhotic livers, and is not specific
Hemochromatosis (HH)
Dz caused by inc Fe absorption (hemosiderosis)
- menstruation helps to delay onset
Primary is AR (85% have HFE gene [C282y] or H63D on cr 6)
- C282Y gives highest iron overload; worse prognosis (but less common); H63D (histidine to aspartate) gives lowest iron overload
-- can be a compound heterozygote c C282Y / H63D intermediate
- other types have mutated HAMP gene (for hepcidin; type 2B), transferrin-receptor-2 gene (type 3) and ferroportin-1 gene (type 4)
HH classification:
Type Gene mutated
• 1 HFE
• 2A hemojuvelin-
• 2B hepcidin
• 3 transferrin receptor2
• 4 ferroportin
Genes: HFE (C282Y, H63D) [6p21.3]
Juvenile hemochromatosis A - HFE2 (hemojuvlin) [1q21]
Juvenile hemochromatosis B - HAMP (hepcidin) [19q13]
Hemochromatosis type 3 - TFR2 (transferrin receptor) [7q22]
Familial iron overload - FTH1 (ferritin heavy chain) [11q13]
Secondary is from chronic transfusion tx (ie in B-thal); asoc c HLA-A3
Sx: Triad of micronodular cirrhosis, diabetes mellitus, and skin pigmentation (from hemosiderin [ferritin breakdown product])
*** Bronze diabetes ***
- may be able to set off metal detectors in airports
Dx: can be confirmed c MRI of abdomen which shows inc Fe deposits in liver; or can do C282y gene testing
- inc ferritin, inc iron, dec TIBC, inc transferrin
DDx: Kupffer cell primary (multiple transfusions); hepatic and Kupffer cell primary from excessive oral iron intake
Tx: repeated phlebotomy, deferoxime
Px: 15% can get HCC
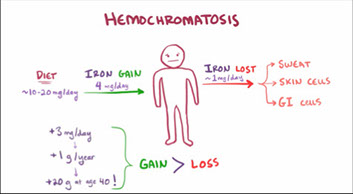


Hemosiderosis



Hemochromatosis

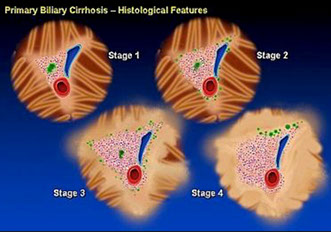
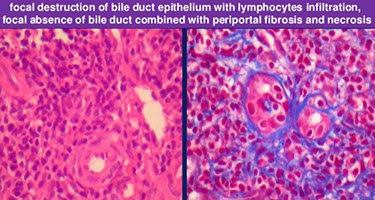
PBC
PBC

PBC


Diagnostic duct destructive lesion
Duct destruction with lymphoplasmacytic infiltrates suggestive of but not necessarily diagnostic of PBC

Stage 2 PBC: cholangiocytes proliferating along bile duct stroma (not a diagnostic lesion)
Primary biliary cirrhosis (PBC)
Autoimmune rxn that destroys medium- to small-sized intrahepatic bile ducts (and not larger!) leading to lymphocytic infiltrates and granuloma formation
- 90% are middle-age females; typically northern European ancestry, c new pruritis and abnormal liver enzymes
- assoc c CREST, RA, celiac dz, Hashimoto, Sjogren syndrome
-- may be part of a larger autoimmune syndrome c destruction of salivary glands and autoimmune gastritis, can also have scleroderma and thyroiditis
- PBC is very patchy, doing bx's on different spots of the liver at the same time will show different stages of dz, may even be able to pick up diagnostic lesions on different levels (should get 6 deeper levels if there is a lesion suspicious for PBC)
- if deeper levels do not work, consider getting CK7, maybe turned on by hepatocytes from reversing the flow of bile from the apical to basal side of cell, but indicates cholestatic liver dz
- CK 7 also stains bile ducts
- CK19 stains canals of Hering, which are dec in early PBC
PBC, stage 1: Bile duct injury
-
PBC, stage 2: Ductular reaction
Late stage PBC, stage 3 to 4: Fibrosis to Cirrhosis
- recurs after liver transplant in 1/3 of pts, after 3-6 years, and looks the same as in non-transplant pts
- should have anti-mitochondrial abs (AMA) +
- florid duct lesions helpful
- may mimic AI clinically, serologically and on micro, but should be managed as AI in these cases to stop fibrosis advancement
Sx: Pruritis, jaundice, dark urine, light stools, HSmegaly; xanthelasma / xanthoma, osteoporosis
Dx: inc conjugated bilirubin, inc cholesterol, inc alk phos, inc serum anti-mitochondrial abs (AMA) directed against pyruvate dehydrogenase complex (PDC), known as M2 fraction or M2 abs (~95%+), IgM inc (hypergammaglobulinemia)
- in biliary cirrhosis in general, can see bile lakes in liver parenchyma on cholangiography
- +ANA in 1/3
- AMA negative PBC sometimes called autoimmune cholangiopathy (may want to repeat AMA testing a second time if strongly suspect)
Micro: 2/5 have granulomas either in portal tracts or lobules
- can have mod to marked inflam of medium-sized portal tracts, c prominent plasma cells and some interface activity, which may be assoc c reactive epithelial changes
- smaller branches of portal tracts c mild nonspecific lymph inflam
- florid duct lesion - duct injury assoc c cuff of histiocytes and granulomatous inflam
- lobules c minimal patchy lobular lymph inflam c occasional apoptotic bodies, and can sometimes have nodular regenerative hyperplasia (NRH)
Small portion may have features that overlap c autoimmune hepatitis, and is called PBC-AIH overlap syndrome
IHC: (+) CK7 hepatocytes indicate chronic cholestasis (also stains normal bile ducts); CK19 stains Canals of Hering, which are decreased in early stages of PBC (stage 0: minimal change PBC)
DDx: drug reaction (even herbal supplements)
- chronic hepatitis (CK19 will show normal canals of Hering)
Tx: ursodeoxycholic acid (UDCA) delays fibrosis progression, cholestyramine (for pruritis)
Px: inc risk of HCC

Secondary Biliary Cirrhosis
Caused by extrahepatic biliary obstruction (stones, chronic pancreatitis) that increases pressure in intrahepatic ducts causing injury/fibrosis and bile stasis
Sx: Pruritis, jaundice, dark urine, light stools, HSmegaly
- may get complicated by hypergammaglobulinemia or ascending cholangitis
Dx: inc conjugated bilirubin, inc cholesterol, inc alk phos
Primary Sclerosing Cholangitis (PSC)
Immune mediated dz mostly in the extrahepatic bile ducts
- periductal fibrosis ("onion skinning") with beading of intra- and extra-hepatic bile ducts on ERCP
- usually in young or middle-aged men (2-3M>1F) and assoc c Ulcerative Colitis spreading up into bile ducts from intestines; also hypergammaglobulinemia (IgM)
- adjacent periportal hepatocytes are swollen (pseudoxanthomatous change) 2/2 salt retention
- nonsuppurative (ie lymphoplasmacytic) destructive cholangitis
can recur in transplanted pts in up to 1/3 of pts, usually after ~5 years
- micro findings same as in non-transplanted pts, though may be worse in recurrent vs native dz
- DDx: biliary stricture or obstruction from surgery or other mechanical causes which can't be differentiated from histo findings, and need help from imaging studies
-- strictures in first 90 days likely procedure related
Sx: Pruritis, jaundice, dark urine, light stools, HSmegaly
- look for middle-aged men c positive P-ANCA (60%)
- sx do not resolve c resolution of IBD, may still need liver transplant after colectomy from UC
Micro: progresses slowly, as dz progresses shows patchy bile ductular prolif c mild mixed portal inflam
- as progresses even more, liver gets scarred, starting in the portal tracts c an irregular outline
- be careful, as onion skinning may be overdiagnosed
IHC: Perioportal copper overload visible c Victoria blue (actually stains an intralysosomal metallothionein protein [copper-binding protein])
Dx: only cause of cirrhosis where bx is NOT the most accurate test - beading on ERCP is the preferred method (may cause secondary cirrhosis)
DDx: infx of extra-hepatic biliary tree is rare, but may occur c Isospora belli
- ischemic cholangitis (usually in setting of liver transplant)
Tx: cholestyramine, UDCA
Px: at risk of developing cholangiocarcinoma (esp if involves major hepatic ducts)
Autoimmune Sclerosing Cholangitis (ASC)
- overlap bwt AIH and PSC, MC in kiddos and young adults
-- autoimmune cholangitis (without "sclerosing") is an AMA-neg PBC
- ASC has typical features of AIH and imaging studies typical of PSC
- UC is strongly linked to both ASC and PSC
PSC

PSC with onion skin fibrosis, by Vikram Vishpande (Twitter)
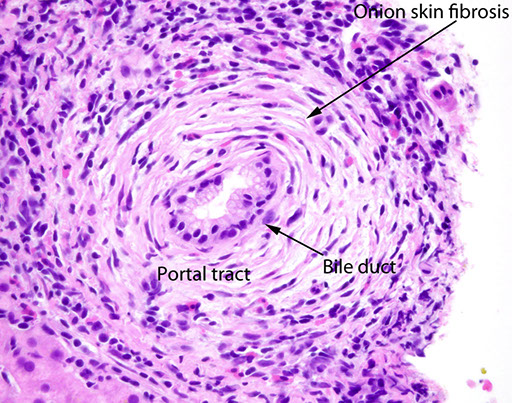
Small Duct Primary Sclerosing Cholangitis
Presents similar to large duct PSC, may also be due to inflammatory bowel dz (UC > Crohn)
Micro: same as large duct PSC
Dx can only be made on liver bx, which requires:
1) liver findings typical of primary sclerosing cholangitis (PSC)
2) normal imaging studies of extrahepatic bile tree
3) exclusion of other causes of biliary tract dz
Px: better than large duct PSC
IgG4 Sclerosing Cholangitis
Chronic autoimmune inflam dz of biliary tree assoc c inc serum IgG4 and inc IgG4+ plasma cells
- M>F, usually ~60 yo, though can first present at 20 yo
- usually occurs c obstructive biliary tract dz (3/4)
- also assoc c type 1 autoimmune pancreatitis (>90%), but not UC
- strictures seen in IgG4 dz are responsive to steroids (not so in regular PSC)
- can also present c hilar mass concerning for cholangiocarcinoma
Micro: can be very similar to PSC, most changes 2/2 large duct strictures that affect the extrahepatic biliary tree
- can see larger portal tracts c mod patchy lymphs enriched for plasma cells, and scattered eos
-- inflam mostly around portal veins can cause a paucicellular halo around the bile duct
- edematous, large inflamed portal tracts cam have irreg stellate profile
- can sometimes see lobular cholestasis and mild lymph lobular hepatitis
- obliteratic phelbitis is a rare finding, but favor IgG4 SC
If presents as a mass, bx shows dense fibrosis c chronic inflam (lymphs and plasma cells), admixed c atrophic cords of hepatocytes and bile ductules
IHC: >10 IgG4+ plasma cells per hpf specific, but not sensitive
- the number of plasma cells usually reflects the size of the bile duct sampled
- even at high levels, IgG4+ plasma cells not specific for IgG4 SC
DDx: overlaps c PSC
- if presents as a mass lesion, consider inflammatory pseudotumor, or an abscess
Sclerosing Cholangitis c Granulocytic Epithelial Lesion
form of sclerosing cholangitis (SC) c granuloar epithelial lesions
- most cases seen in kiddos c inflam bowel dz
- cholangiogram shows diffuse abnormalities in intrahepatic bile ducts, though extrahepatic bile ducts usually spared
- serum ANA can be +
- most show considerable clinical and histologic overlap c autoimmune sclerosing cholangitis (differentiated by presence of granulocytic epithelial lesion)
Micro: bile duct c extensive neuts infiltration of duct epithelium
Caroli disease
Marked bile duct dilation or macroscopic cysts (centimeter range) visible on imaging
- Caroli syndrome has hepatic fibrosis in addition visible cysts
- predisposes to acute bacterial cholangitis and recurrent stone formation
- Caroli disease and Caroli syndrome are probably a spectrum of the same dz
- assoc c ARPKD, nephrolithiasis, and PKHD mutations [6p]
Caroli dz - intrahepatic bile duct dilation

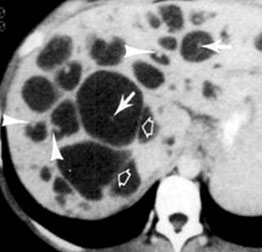

Congenital hepatic fibrosis
Enlargement of portal tracts by intestinal fibrosis within which there are segmentally dilated and abnormally shaped (sleevelike) bile ducts; complicated by portal hypertension
- dx made by liver bx
- is a form of ARPKD and presents in kiddos and young adults
Micro: fibrosis usually not present at birth, but can progress to cirrhosis, also severe chronic inflam (+/- acute inflam), luminal inspissated mucin, and bile
- may have intrahepatic cysts that are connected to biliary tree
Genes: mutations in PKHD1 (codes for fibrocystin involved in embryogenesis of the tubules in liver and kidne)
Polycystic Liver Disease (PCLD)
- rare autosomal dominant disease of the liver and should not be confused with the autosomal recessive associated fibropolycystic liver diseases, discussed later. The genetic alterations in PCLD affect PKD1 or PKD2 and PRKCSH or SEC63 genes. Germline mutation in PKD1 or PKD2 occur in patients with autosomal dominant polycystic kidney disease (AD-PKD). PKD1 and PKD2 are located, respectively, on chromosomes 16p13.3 and 4q21–23 and encode polycystin-1 and polycystin-2. The incidence of AD-PKD ranges from 1 in 400 to 1 in 1000 and accounts for roughly 80% to 90% of all patients with PCLD. In patients with mutations in PRKCSH (which encodes hepatocystin) or SEC63 (which encodes Sec63 protein) genes on chromosome 19p or 6q21, respectively, PCLD occurs in isolation without PKD. These isolated PCLD are estimated to affect 1 to 10 cases per 1000000 individuals. The existence of chronic renal failure in the presented patient would therefore make it important to rule out underlying PKD1 or PKD2 mutation
- a female preponderance at a ratio of 6:1, leading to postulation that estrogen plays a role in cyst development
- majority of AD-PKD patients have an underlying PKD1 or PKD2 mutation, chronic renal failure due to polycystic kidneys is commonly the presenting feature
- PCLD belongs to a wider group of diseases referred to as fibropolycystic diseases. This group of diseases share underlying genetic abnormalities of epithelial cilia function and development, making them “ciliopathies.” Some of the other members of the group include congenital hepatic fibrosis (CHF), Caroli disease/Caroli syndrome (CD/CS), and choledochal cyst. PKHD1, present on chromosome 6p21.1p12, encodes fibrocystin/polyductin and accounts for most cases of CHF, CD/CS, and autosomal recessive PKD (AR-PKD)
Micro: cysts lined by cuboidal to flattened nonmucinous biliary-type
epithelial cells surrounded by fibrous stroma. Dysplasia and malignancy are absent.
- Bile, with or without luminal calculi, can be seen in some sections. The interspersed hepatic parenchyma shows varying features including chronic compression, complete parenchymal loss, fibrosis with ductular reaction, and relative preservation with dilated sinusoids. Some sections show luminal hemorrhages, as well as scattered von Meyenburg complexes
Px: PCLD is a progressive disease that presents with increasing number and diameter of cysts, and subsequently increasing overall liver size throughout life. By the age of 70 years, 75% of affected patients will have developed clinically apparent cysts
- Chronic liver failure occurs with time due to progressive compression and fibrosis of the liver parenchyma and is characterized by increasing bilirubin, elevated international normalized ratio (INR), low albumin, worsening ascites, and upper gastrointestinal varices. Treatment options include reducing liver mass through abdominal percutaneous puncture, laparoscopic or open fenestration, or surgical resection, but transplantation is currently the only curative option. Long-term outcome for transplantation after the first two months is excellen
PCLD

Biliary (Choledochal) cysts
MCC congenital extrahepatic bile duct dilation, also seen in intrahepatic biliary radicles or both, 4F>1M
- more prevalent in Asia than USA
- Originally called "choledochal" but later revised to include intrahepatic cysts within the biliary system
- triad of sx: pain, jaundice, and mass in right upper quadrant
5 types:
Type 1: (up to 9/10) cystic / fusiform Bile Duck (BD) dilation, but cannot be intrahepatic, has A, B and C subcategories
1A - CBD or adjacent duct, assoc c Abnormal PancreatoBiliary Junction (APBJ)
1B - focal/segmental bile duct dilation outside liver (not assoc c APBJ)
1C: diffuse smooth fusiform (not cystic) dilation of extrahepatic BDs, assoc c APBJ
Type 2: (1/50) BD diverticula, may pop up in any part of BD system
Type 3: (1/20) aka choledochoceles, cysts coming from part of BD in the duodenum (?), can thus be lined by BD or duodenal epithel?
- may come from duplications of the ampulla into duodenum
- 2+ subtypes based on anatomical features
Type 4: (up to 3/10) anywhere in BD system, see multiple cysts
- divided into subtypes based on cyst location
Type 5: (1/5) aka Caroli disease, multiple cystic dilations in liver BD
Dx: first suggested by US or CT, MRI then preferred
- ERCP can be done on some subtypes to see if blocked BDs
Tx: Roux-enY hepaticojejunostomy
Px: assoc c inc risk of cholangioca (esp in types I-4); slightly higher risk after excision
Abnormal PancreatoBiliary Junction (ABPJ)
- aka pancreatobiliary maljunction, may be slightly more prevalent in Asian countries, assoc c bile duct cysts
- pancreatic duct fuses c BDs outside duodenal wall, causing formation of a really long common duct channel (ampulla of Vater is small or flat)
Physiologic neonatal jaundice
Immature UDP-glucoronyl transferase at birth unable to conjugate bilirubin, leading to jaundice, which may lead to kernicterus
Tx: Phototherapy (converts bile to a H20 soluble form for secretion)

Schematic of bilirubin (B) metabolism in the hepatocyte. Glucuronicacid (G), bilirubin glucuronate (BG), uridine diphosphate glucuronic acid (UDPGA), Glutathione-S-transferase (GST), multidrug resistance protein 2 (MRP2), UDP-glucuronyltransferase (UGT1A1-specific isoform)
Dubin-Johnson syndrome (DJS)
Bilirubin excretion disorders
*** Get Closer, Deeper Romance ***
Conjugation disorders: Gilbert and Criggler-Najjar
Secretion disorders: Dubin-Johnson, Rotor syndrome
Gilbert's syndrome
AR c underactive bilirubin-UGT (mutations in the 5' TATA box of the UGT1A1 promoter) causing prob c bilirubin uptake leading to unconjugated bilirubinemia
- No overt hemolysis
- Assoc c stress (fasting, viral illness)
- otherwise asymptomatic; may be present in up to 5% of whites
No tx necessary (avoid stress :) )
Criggler-Najjar syndrome
Type I: Absent UDP-glucoronyl transferase (UGT1A1) causes unconjugated bilirubinemia (due to absent bilirubin conjugation), which is fatal in first few years of life
- absent conjugated bili
- negative urine bilirubin
- dec urine bilinogen
- light-brown poop
Type II (Arias syndrome): less severe (UDP-G just decreased, not absent and can be tx'd c phenobarbitol
Sx: jaundice, kernicterus, inc unconjugated bilirubin
Tx: plasmaphoresis, phototx
Dubin-Johnson syndrome
AR, defective canalicular multiple organic anion transporter (cMOAT) in ABCC2 gene
- prob c conjugated bilirubin excretion leading to conjugated hyperbilirubinemia
Liver appears grossly black
B9
Rotor's syndrome is similar to Dubin-Johnson but is milder and does not cause a black liver
Dubin-Johnson syndrome


Rotor Syndrome
AR; multiple defects in hepatocellular uptake and excretion of bilirubin pigments, gene unknown
Sx: asymptomatic conjugated hyperbilirubinemia, jaundice, normal lives; liver looks normal
Banff Liver Rejection Schema
Rejection Activity Index (RAI)
Portal Inflammation
1- lymph in a few of the portal triads (which aren't expanded)
2- Majority of portal triad expansion c mixed inflammatory infiltrate (lymphs, neutros, eos, blasts)
3- Marked expansion of majority of portal triads c mixed infiltrate that goes into periportal parenchyma
Bile Duct Inflammation
1- Minority of bile ducts cuffed and infiltrated by inflam infiltrate c mild epithelial reactive changes (inc N:C)
2- Majority of bile ducts inflamed c occasional degenerative changes (nuclear pleomorphism, disordered, cytoplasmic vacuolization)
3- Same as 2, but higher amt of degenerative changes and focal lumen disruption
Venous Endothelial Inflammation
1- Subendothelial lymph inflam c minority of portal / hepatic venules
2- Same as 1, but majority of portal / hepatic venules
3- Same as 2, but with perivenular inflam extending into parenchyma c perivenular hepatocyte necrosis
Acute rejection
MCC liver problems in 1st year after transplant; MCC liver allograft lesion
Triad:
1) Inflam c mostly lymphs, but also some eos
2) Bile duct damage
3) Portal vein endotheliitis
- inflam of central vein can be present in more severe cases
Microabscesses seen after transplantation are 2/2 CMV hepatitis
Herpes hepatitis leads to vast geographic necrosis
Q fever
Rickettsial (Coxiella burnetti)
Target of fibrin-ring granuloma ("donut granulomas")
- central fat vacuole
- layer of histiocytes and lymphocytes
- a fibrin ring
DDx: CMV, HAV, allopurinol tox, Leishmaniasis
Q fever

Bile Duct Adenoma
- aka peribiliary gland hamartoma
B9 aggregate of small tubular bile duct-like structures w/o lumens or bile, growing in variably fibrous stroma; usually in cirrhotic livers
- possibly a type of pyloric gland metaplasia of the peribiliary glands of the liver, some consider it a neoplastic lesion
- may arise from peribiliary glands
Grossly are similar to bile duct hamartomas, as a small (<1 cm) lesion under the liver capsule, usually single lesion
Micro: medium-sized regular bile duct structures (no prominent nucleoli or pleomorphism) in a densely hyalinized fibrous stroma
- bile ducts do not have a lumen, lack bile, and w/o anastomoses
- cells have consistent nuclear size, no nucleoli
- sometimes can look oncocytic
- usually but can have somewhat infiltrative borders
-usually mild inc lymphs
- see lymph aggs at junction bwt normal liver and adenoma
IHC: (+) MUC5AC/6, acid mucin, bile ducts stain as usual
DDx: focal fibrosis, mets, healed granuloma, von Meyenburg complex, FNH, hemangioma, AV malformation, well-diff cholangiocarcinoma (these would have prominent nucleoli, complex glands, mits, luminal necrosis; may be impossible to differentiate on needle bx)
Px: Rare reports of them turning malig, but generally no malig potential
Clear Cell Bile Duct Adenoma
Rare subtype of bile duct adenoma, c cells having diffuse clear cell change; cells grow in small nests and cords c round to oval nuclei w/o much pleomorphism
- low prolif rate by Ki67, border of lesion less well defined than traditional adenoma
- should not have inc mits, no necrosis, no atypia (vs carcinoma)
- usually c mild inc lymphs
DDx: clear cell cholangiocarcinoma and met clear cell ca

Bile duct adenomas are well circumscribed but unencapsulated. Note the fibrous stroma and evenly distributed glands. An adjacent normal portal tract (arrow) is present.

Some bile duct adenomas have eosinophilic cytoplasmic globules composed of α-1-antitrypsin (arrow). Note the lack of nuclear atypia, mitoses, or nuclear hyperchromasia in the lining epithelium of small rounded glands.
Biliary Adenofibroma
Rare biliary tumor made of dilated tubulocystic structures in a dense fibrous stroma; up to 2 cm
- recently described, may previously have been called con Meyenburg complex of bile duct adenoma
- biliary structures larger than in adenoma or von Meyenburg complex and can be filled c blood or proteinaceous material or bile
- glandular structures can be serpentine and anastomosing in some parts and look like von Meyenburg complexes
IHC: epithelial cells CK7/19 +; stromal cells are myoblastic and vimentin and smooth muscle actin +

Biliary adenofibroma. (a) Tubular structures with fibrous stroma characterize this tumor. Focally, micropapillary features are found in the left lower corner. (b) Papillary features with complicated structures suggest an imminent malignant transformation. HE.
Simple Cysts
- aka solitary bile duct cysts
Solitary cyst lined by simple cuboidal epithelium and lacking ovarian stroma; 8F>>1M, ~50 yo, most are asx
Gross: usually single cyst but can be multiple smaller cysts
Micro: lined by simple biliary-type epithelium on dense layer of fibrous tissue
- epithelium may have intestinal, pyloric, or squamous metaplasia, and sometimes be somewhat dysplastic
- epithelium may be sloughed off and difficult to find
- if arises from hilum of near big intrahepatic portal tract it would be a peribiliary gland cyst
- the wall can have small aggregates of bile ducts or peribiliary type glands
- may have inc macrophages if cyst has ruptured
DDx: mesothelial cyst (small and subcapsular, lined by mesothelial cells [not biliary type cells], calretinin, WT1, CAM5.2, AE1/3, and EMA +)
- foregut cyst (also subcapsular, lining has cilia, and smooth muscle bundles seen in walls of the cyst)
Bile Duct Hamartoma
- aka biliary hamartoma, bile Duct plate malformation, von Meyenburg complex
Common finding in needle bx, explant and postmortem liver
- B9, small lesion of anastomotic bile ducts
- usually acquired lesion, more common in cirrhotic livers, esp c HCV and EtOH damage (risk factors similar to those for intrahepatic cholangiocarcinoma)
Gross: <1 cm white nodule underneath liver capsule (like bile duct adenoma), can be single or multiple
- sometimes can be larger and have large dilated biliary cysts and lack ovarian stroma (known as a giant cystic bile duct hamartoma)
Caused by normal remodeling of the bile duct plate, results in dilation and irregular contours of bile ducts
Micro: aggregate of bile ducts c open lumens c bile that grow in an irreg and anastomosing way; can have lots of myxoid or fibrotic stroma
IHC: will stain similar to normal bile ducts, have low to absent Ki67
DDx: bile duct adenoma (also a small white nodule underneath liver capsule, have small or absent lumens, no bile, grow in small gland-like structures)
Px: malig potential very low, but is still there
- may see cholangiocarcinoma arising from hamartoma
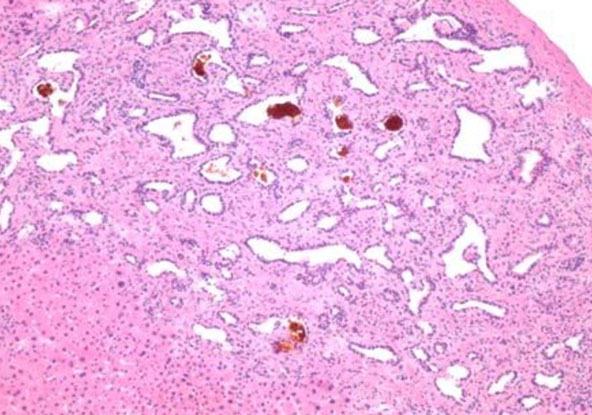
Bile duct hamartomas (von Myenberg complexes) are potential mimickers of metastatic adenocarcinoma. They have the three features seen in this case: 1) there are multiple lesions; 2) they contain bile, 3) they are lined by a flattened epithelium. Bile duct adenomas are usually solitary and they do not contain bile. Pleomorphism and an invasive growth pattern help distinguish adenocarcima from bile duct adenomas and hamartomas
Adenomyomatous Hyperplasia
- aka adenomyosis of the gallbladder
Common lesion of the gallbladder, considered a form of diverticular dz
- invaginations of the mucosa with smooth muscle hyperplasia
Micro: variably dilated glandular elements, surrounded by bundles of smooth muscle
- branching duct or gland-like structures in the wall of the gallbladder and hyperplasia of smooth muscle cells
- epithelial lining is single layer of tall columnar cells
- can occasionally have perineural invasion, which does NOT necessarily indicate malignancy
DDx: invasive adenocarcinoma (similar histologically)
Px: not considered premalig


Mucinous Cystic Neoplasm
- aka biliary cystadenoma, hepatobiliary cystadenoma, or (if malig) cystadenomcarcinoma
Cyst lined c biliary type epithelium c ovarian type stroma in wall
- can be 1) low- or intermediate-grade neoplasia, 2) high-grade neoplasia, 3) mucinous cystic neoplasm c invasive carcinoma
- similar to mucinous cystic neoplasm of pancreas
- 20F>>1M, usually in 30-40 yo
- usually solitary and multilocular; fluid content usually thin and clear, though can be mucinous, bloody or purulent
- should not communicate c bile ducts, bc then it would be an intraductal papillary neoplasm c marked cystic change
Micro: lined by columnar epithelial cells c basal nuclei and apical mucin; but can be cuboidal or flat
- intestinal meatplasia common, pylori gland metaplasia and squamous metaplasia also possible
- may not have mucinous epithelium, but must have ovarian stroma in that case
- epithelium can have low- or high-grade dysplasia, which can be from crypt-like invaginations of epithelium to cyst wall, micropapillary projections, epithelial multilayering, hyperchromasia, nuclear variability
-- High-grade dysplasia (formerly carcinoma in situ) has marked papillary projections, c complex tubulopapillary architecture, inc cellularity, lots of mits, inc nuclear pleomorphism (should sample for invasive carcinoma)
IHC: (+) CK7/19/8/18 in epithelium; scattered endocrine cells are SYN/CHR +, ovariant stroma is (+) for ER/PR and a-inhibin
- neg CK20, except in dysplastic areas
Intraductal Papillary Biliary Neoplasm (IPBN)
- aka intraductal papillary neoplasm, biliary papillomatosis, biliary papilloma
Papillary tumor growing in biliary tree that can cause focal cystic change, are 1/10 bile duct cancers, and is the bile duct counterpart of intraductal papillary mucinous neoplasm (IPMN) of pancreas
- M=F; hx of bile stones usually, most are asymptomatic, but can show biliary obstruction
Gross: large single cyst c multiple subdivisions made by thin fibrous walls that connects to bile ducts (must gross carefully to document this!!!!)
Micro: epithelial cells in duct lumen c micropapillary or complex tubulovillous architecture
- 1/3 have mucin production
- may dilate the bile duct a lot and mimic mucinous cystic neoplasms (use ovarian stroma and bile tree connection to differentiate)
- epithelium can show range of morphologies, such as biliary type (pancreatobiliary - looks like dysplastic biliary epithelium), intestinal type (epithelium made of tall, thin columnar cells c basal nuclei that looks like intestinal epithelium); gastric type (epithelium c tall round cells c lots of mucin that has a clear appearance); oncocytic type (epithelium has pink cytoplasm)
-- biliary tree epithelium is the MC
-- if ca develops, intestinal type epithelium assoc c colloid ca, and biliary type epithelium assoc c tubular AC
- important to submit the whole tumor to exclude invasive component
Px: high risk of invasive ca with these lesions, so must be grossed well
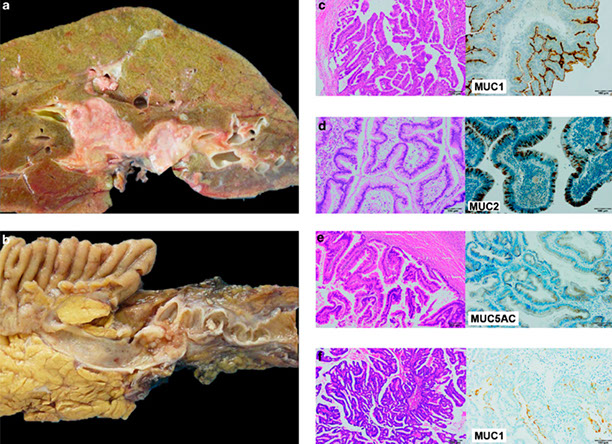
Pathology of intraductal papillary neoplasms of the bile duct. (a) Intrahepatic intraductal papillary neoplasm of the bile duct showing dilated biliary ducts filled with tumor tissue. (b) Extrahepatic intraductal papillary neoplasm of the bile duct with papillary tumor tissue in the ductus choledochus. (c) Pancreato-biliary subtype with diffuse expression of MUC1. (d) Intestinal subtype with diffuse expression of MUC2. (e) Gastric subtype with expression of MUC5AC. (f) Oncocytic subtype with focal expression of MUC1.
Cholangiocarcinoma
Divided into Intrahepatic and Extrahepatic
- Klatskin tumors are those coming from the right or left hepatic duct, or at the junction of the hepatic ducts
- hilar cholangiocarcinoma is a synonym for a Klatskin tumor, though not used consistently
- cholangiocarcinoma coming from the Intrahepatic right/left bile ducts called perihilar cholangiocarcinoma, but not always used consistently
- precise location of larger tumors may not be clearly intra- or extrahepatic, and may never know for sure
Risk factors: Clonorchis sinensis, Ca2+ bilirubinate stones, fibrocystic disease, Caroli's disease, PSC / UC, EBV, thorotrast
- most cases of intrahepatic cholangiocarcinoma (ICC) occur in non-cirrhotic livers
Precursor lesions are biliary intraepithelial neoplasia (BilIN), which is divided into BilIN-1, -2, and -3
- BilIN-2 and -3 are considered high grade and found in cirrhotic livers in med- and large branches of the bile tree and assoc c HCV and EtOH-assoc liver dz
-BilIN-1 is difficult to separate from reactive changes
Micro: has variety of growth patterns, such as irreg branching tubular structures or irreg aggs of infiltrating glands
- do not always have a clear lumen and can grow in solid trabeculae and nest mimicking HCC
- may have a colloid morphology
- more centrally located lesions more likely have columnar type epithelium, well-formed glands, and make mucin
- more peripheral lesions have irreg anastomosing, tubular structures c low cuboidal cells that do not make mucin and look like proliferating ductules, and is called bile ductular pattern; these are more likely to not have a lumen and grow as solid nests and trabeculae
- rarely, may have clear cell morphology and sarcomatoid growth
Usually c dense desmoplastic type fibrotic response, sometimes c stromal elastosis
- may colonize and extend along bile duct epithelium (not specific, can be seen in mets from colon)
- can grow along portal tracts by growing into connective tissue, w/o direct invovement of bile duct (dirty luminal necrosis and high Ki67 can help you to make the dx)
IHC: (+): mCEA, pCEA (cytoplasmic), CK7/20, CK19 (up to 10/10) AE1/AE3, Cam5.2, EMA, MOC31, Mucicarmine
- neg: HepPar-1, aFP, CD10/villin, Glypican-3 and Arginase-1
- nothing is really specific for cholangioca, and is a dx of exclusion

Iintrahepatic, cholangiocarcinoma (right of image); benign hepatocytes are seen (left of image). Histologically, this is a cholangiocarcinoma as (1) atypical bile duct-like cells (left of image) extend from the tumor in an interlobular septum (the normal anatomical location of bile ducts), and (2) the tumor has the abundant desmoplastic stroma often seen in cholangiocarcinomas. A portal triad (upper-left of image) has a histologically normal bile duct. H&E stain.
Cholangiolocarcinoma
Considered a better-acting version of cholangiocarcinoma
- dx made on H&E
IHC:
GenesL likely a separate entity; cluster together
- have upregulated biliary specific genes
- positive NCAM (CD56) / EpCAM
Px: Better

Cholangiolocarcinoma
Lymphoepithelioma-Like Cholangiocarcinoma (LELC)
- aka lymphoepithelioma-like carcinoma,
Tumors c irreg sheets of poorly define epithelial cells in a dense lymph infiltrate
- very rare, may be MC in Asian population
Micro: attenuated epithelial structures c poorly defined sheets of epithelial cells in dense lymph background
- probably has areas of more typical cholangiocarcinoma in resection specimens
IHC: (+) CK7/19, EBV-ISH
- neg Hep-Par
Px: not clear bc really rare, may have a somewhat better px
LELC. A: The tumor is well circumscribed with a thick fibrous capsule (HE stain, × 20); B: The majority of the tumor is composed of well differentiated glandular cells with dense lymphocytic infiltration and scattered lymphoid follicles (HE stain, × 200); high-power view of another area in the tumor; C: The tumor reveals a solid, poorly differentiated growth pattern (HE stain, × 200); D: In situ hybridization for Epstein-Barr virus-encoded RNA is diffusely positive in the tumor cells (× 200). [2]

References
1. Lin et al. Fibrolamellar carcinoma: A concise review. Arch Pathol Lab med, 2018.
2. Liao TC. Lymphoepithelioma-like cholangiocarcinoma: A mimic of hepatocellular carcinoma on imaging features. World J Gastroenterol 2015, April 7; 21(13):4089-4095
3. Shetty S. Giant-cell hepatitis- Rare entity in Adults. J Clin Exp Hepatol. 2016 Sep; 6(3):244-245.