Lipoproteins
Introduction
Type I: Hyperchylomicronemia
Type II: Hyperbeta-lipoproteinemia
Type III: Dysbeta-lipoproteinemia
Type IIb / IV: Familial Combined Hyperlipidemia
Type V: Mixed type lipoproteinemia
Familial hyperalphalipoproteinemia
Hypoalpha-lipoproteinemia
Anaplha-lipoproteinemia (Tangier disease)
Hypobeta-lipoproteinemia
Abeta-lipoproteinemia
Introduction
RR studies for cholesterol established
250 – 280 mg/dL
40 – 50% develop CHD
National Cholesterol Education Program Adult Treatment Panel III (ATPIII)
All adults (20 yrs and older) have fasting lipid profile every 5 years
Total cholesterol (TC) - High >240; Desire <200
Triglycerides (Tgs) - see below
LDLc - High 160-189; Desire <100
HDLc - Low <40, High > 60
NCEP guidelines (triglycerides)
Upper limit of normal 150 mg/dL
Borderline = 150 – 199 mg/dL
High = 200 – 499 mg/dL
Very high = ≥500 mg/dL
Apo-A
A-I –primary protein component of HDL
- activates LCAT: HDL receptor binding to allow
cholesterol extraction from cells
A-II –not athero-protective
Apo-B
Several forms in circulation
Key risk factor for developing atherosclerosis
B48 - found in chylomicrons; provides structural support
B100 - found in LDL and VLDL; Structural support; receptor binding to deliver cholesterol to target cells; feedback inhibition of liver cholesterol synthesis
Apo-C
Required for the metabolism of chylomicronsand VLDL
CII - found in chylomicrons, VLDL; early (nascent) HDL molecules; activates lipoprotein lipase
Apo-E
Found in Chylomicrons, VLDL, IDL; some HDL molecules; Receptor binding to clear lipoprotein remnants from circulation
- Varying genotypes influence cholesterol level
- MC is ApoE3
- ApoE4 high binding, high LDL, also linked to Alzheimer dz
- ApoE2 low binding, causes high IDL (famlial dyslipidemia typie III)
Apoprotein (a)
Binds to LDL; homology with plasminogen; may
inhibit clot lysis, facilitate cholesterol uptake by
scavenger receptors
Cholesterol
Synthesis in liver stimulated by free fatty acids, esp unsaturated fatty acids;
- dietary intake minor factor affecting cholesterol levels.
- cholesterol synthesis begins with acetyl CoA, and HMG-CoA reductase is rate limiting step (inhibited by the various statins)
- Cholesterol is needed for membrane structure
and for steroid hormone synthesis.
Lab: measured using cholesterol oxidase enzyme
Triglycerides
come from diet and liver synthesis
- triglycerides must be cleaved (by lipases, including lipoprotein lipase in tissue) to free fatty acids and glycerol in order to across cell membranes
- important as energy stores; when insulin and/or glucose levels are low, triglycerides are cleaved and free fatty acids are used for energy
Lab: after cleavage by lipase, glycerol measured in one of several ways (MC uses glycerol kinase to produce glycerol-3- phosphate, which is eventually metabolized by other enzymes to produce NADH)
- may be overestimated by endogenous sources of glycerol
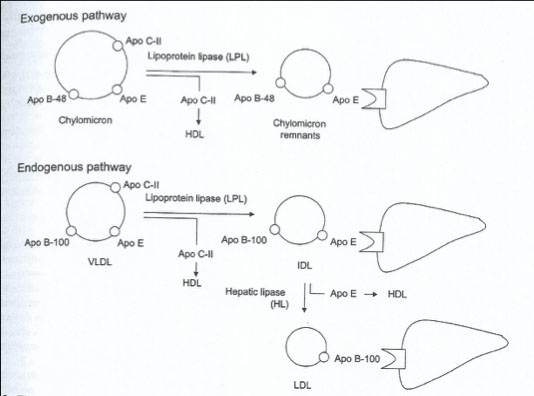
Chylomicrons
Made exclusively in intestine
Contain cholesterol, triglycerides and specific apolipoproteins (B-48, A-1, C, E)
*** Bust 48 chylomicrons out of the gut!!!***
***E = Empty into liver***
Function: to transport dietary triglycerides
Not found in the blood of healthy people
If present, serum is turbid or milky
Floats on top of serum upon standing and refrigeration
Appear at application point (origin) on electrophoreis
Very Low Density Lipoprotein (VLDL)
Composed mostly of triglyceride
52% triglyceride, 22% cholesterol, 18% phospholipid, 8% protein
- migrates to pre-B region of electrophoresis
Two sources:
Dietary –exogenous triglycerides
Hepatic –endogenous triglycerides
Apo-C is the major apoprotein, also has B-100 and E
Not considered atherogenic
Caution: Triglyceride values above 200 mg/dl
Turbid serum a quick and dirty test for evaluation
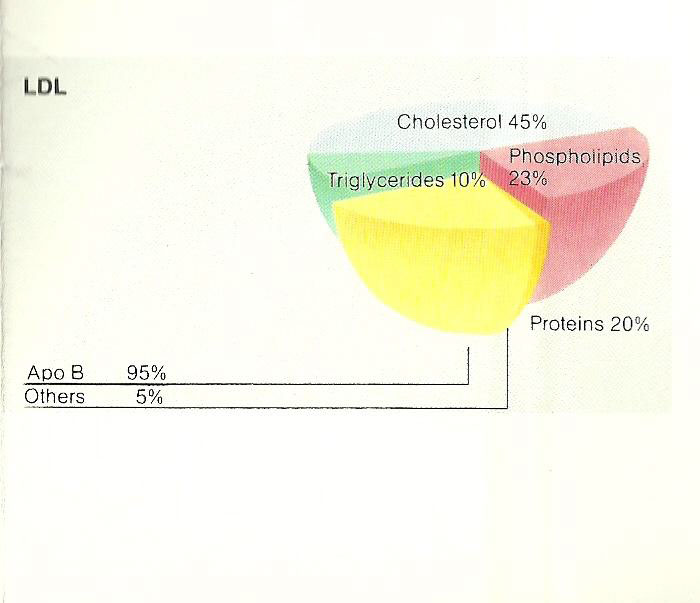
Low Density Lipoprotein (LDL)
Forms through catabolism of VLDL
Friedwald equation
LDL = total cholesterol - HDL - TG/5
Delivers cholesterol to liver or other tissues where it is stored, deposited, or excreted
Composed of 80% lipid and 20% protein
Apo-B-100 is the major apoprotein
- migrates to B area on electrophoresis
Considered the major atherogenic lipoprotein
Two classes exist based on flotation density:
LDL 1 –more lipid rich
LDL 2 –more dense
Small dense fractions indicate high risk for CHD
Labs: MC calculation uses Friedewald equation:
LDL-C = Total Cholesterol – HDL-C – (Triglycerides/5)
- the term TG/5 estimates the VLDL cholesterol, and assumes that no chylomicrons are present, so need a fasting specimen
High Density Lipoprotein (HDL)
Major role is to esterifycholesterol
Many subfractions
HDL2 –
- high levels associated with longevity
- Low levels associated with CHD
HDL3 –associated with severity of CHD
Therapeutic agent in reducing atherosclerotic CVD
Labs: recently, “direct HDL-C” methods available using abs to apo B and apo E
- inaccurate with high triglycerides
Intermediate Density Lipoprotein (IDL)
Density characteristic of VLDL
Associated with “broad-βdisease”
Very atherogenic lipoprotein
Not seen in normal individuals
Lp(a)
Composed mostly of Apo-B
Plays a role in thrombogenesis
Plays a role in atherogenesis
Associated with elevated risk for CHD
Elevations often seen in
Levels can only be lowered with nicotinic acid
Type IIb or Type IV: Familial Combined Hyperlipidemia
Sx: Familial combined hyperlipidemia has been previously classified as type IIb or type IV hyperlipidemia but appears to constitute a specific subgroup. It is, in fact, probably the most common lipoprotein disturbance encountered in clinical practice, affecting possibly 30% of patients who survive myocardial infarction.
- Present with either an increase in LDL or VLDL or both, and in some patients the phenotypical expression may fluctuate depending on diet, alcohol exercise, and body weight. Although cholesterol and Apo B invariably remain elevated, the triglyceride (VLDL) fluctuation may make the diagnostic differentiation from secondary causes problematic
Sx: The major diagnostic feature of familial combined hyperlipidemia is an elevation of Apo B, which is often disproportionate to the cholesterol or LDL-C elevation. Most typically, total cholesterol and triglyceride levels are increased, with the total cholesterol in the 200 to 300 mg/dL range and triglyceride in the 200 to 500 mg/dL range. HDL-C level is often low
Familial combined hyperlipidemia is probably the result of excess hepatic lipoprotein production, with only minimal impairment of catabolic processes for
VLDL remodeling and LDL clearance
Type V: Mixed Lipoproteinemia
likely to be secondary to one of a number of
other disease states. Also, in a given patient, the disorder can fluctuate between type IV and V, which essentially demonstrates the absence or presence of concomitant chylomicronemia superimposed on a marked increase of VLDL values. Neither the exact genetic mechanism for familial type V disorder nor its genetic relationship to familial type IV disorder is understood.
Mech: characterized by elevation of triglyceride levels due to increases in both VLDL and chylomicrons. The increase in chylomicrons can be seen as a creamy surface layer when a plasma specimen is allowed to stand for 16 hours at 4 °C. The turbid to lactescent layer below the surface layer is a result of the VLDL increase. Plasma cholesterol level may be slightly to moderately increase. LDL-C and HDL-C levels are usually normal to low. In some patients, reduction of LPL has been reported.
The presence of circulating chylomicrons and VLDL in fasting plasma indicates that normal clearing mechanisms for triglyceride-rich lipoproteins are inadequate. Postheparin LPL activity is usually normal, which suggests that the defect is not
the same as in type I hyperlipoproteinemia
Familial Hyperalphalipoproteinemia
It appears to be transmitted as an autosomal dominant trait associated with a less than average cardiovascular disease risk.
Longevity analyses demonstrate an 8- to 12-year prolongation of life expectancy for kindred members. Myocardial infarction morbidity and
mortality are reduced.
Secondary hyperalphalipoproteinemia associated with use of estrogenic preparations or alcohol has been described. Elevated HDL-C levels are rarely the
result of estrogen, oral contraceptive, or excessive alcohol intake, but these factors must be ruled out
Distinctive primary elevations of HDL-C levels, slight elevation of total cholesterol level, and normal triglyceride concentrations characterize the syndrome.
Apo A-I is massively increased and are responsible for protection against atherosclerosis. HDL particles containing Apo A-I and Apo A-II.
To avoid a diagnosis of hypercholesterolemia based on total cholesterol values, HDL-C levels should routinely be measured whenever patients' lipid and
lipoprotein levels are assessed. In assigning risk and making therapeutic decisions, a level of 35 mg/dL is the currently recommended cut-off point
No hypothesis for the increased HDL levels found in familial hyperalphalipoproteinemia has been set forth. Although it may be associated with enhanced triglyceride-rich lipoprotein clearance and LPL activity, it is more likely to be due to increased Apo A-I synthesis
Hypoalpha-lipoproteinemia
Studies have shown a significant increase in atherosclerosis risk in persons with low HDL-C levels. Because the differences in HDL-C levels between various risk groups are small (5 -10 mg/dL), HDL-C measurements may be replaced by other measurements of the HDL moiety, such as measurement of Apo A-I.
A number of underlying disease states or environmental influences may be associated with reduced levels of HDL; one of the most common is hypertriglyceridemia. A number of studies have suggested that familial hypo-alphalipoproteinemia follows Mendelian dominant transmission
Ingestion of certain drugs, lack of exercise, obesity, and alcohol consumption are factors that interact with the underlying genetic pattern controlling HDL levels. Identification of individuals with familial hypoalphalipoproteinemia is important in the treatment of other associated risk factors. The underlying defect appears to be in the primary structure of Apo A-I. In another disorder, known as fish-eye disease, HDL levels are reduced to approximately 10% of normal, with an increase in triglyceride levels. Severe corneal opacity is noted clinically. Although the HDL levels are diminished, their apolipoprotein composition appears normal. Atherosclerosis is present in some patients, but it is usually a late manifestation.
Analpha-lipoproteinemia (Tangier disease)
The best-known disorder of HDL metabolism is Tangier disease. This rare autosomal recessive disorder is characterized by accumulation of cholesterol esters in the reticuloendothelial system and in other tissues. The disorder is associated with the virtual absence of HDL in plasma. Homozygous persons have undetectable amounts of HDL-C and extremely low levels of Apo A-I and A-II. Plasma total and LDL-C concentrations are usually low, and mild hypertriglyceridemia is often observed. The disorder is relatively benign because the lipid accumulation in the reticuloendothelial tissue does not usually affect any organ function. Unlike findings in LDL abnormalities, the abnormality in Tangier disease is predominantly cerebrovascular in character.
Hypobeta-lipoproteinemia
more common than previously suspected, probably because of the limited attention focused on the persons in the lower fifth percentile of plasma total cholesterol and LDL-C level distributions. Familial hypobetalipoproteinemia is inherited as an autosomal dominant trait. Like increased HDL-C, reduced plasma LDL-C concentration is associated with significant increases in life expectancy in both men and women, which is probably related to relative diminution myocardial infarctions.
The syndrome is arbitrarily defined as an LDL-C level below the fifth percentile for a given sex- and age-matched population. The condition should not be confu-sed with abetalipoproteinemia. In hypobetalipoproteinemia, LDL-C is always detectable, whereas levels of other lipoproteins such as VLDL and HDL may be, low, normal, or even increased.
Studies suggest that these patients synthesize LDL at only half the normal rate, but that the catabolic rate remains normal. Because LDL is the breakdown product VLDL, patients also have low triglyceride values
Abeta-lipoproteinemia
A rare autosomal recessive disorder characterized by absence of lipoprotein species containing Apo B. This is manifested in the total absence LDL with consequent very low plasma levels of total cholesterol. Persons who are heterozygous for the disorder have no known clinical or biochemical abnormality but may present with hypobetalipoproteinemia. Persons who are homozygous have no detectable chylomicrons, VLDL, or LDL; therefore, levels of triglyceride, cholesterol, and phospholipids are extremely low. Total plasma cholesterol concentrations are usually less than 50 mg/dL, and virtually all of the total plasma cholesterol is associated with HDL. All of the other apolipoproteins, A-I, A-II, and the C series, are usually present
Sx: Malabsorption of dietary fat causes poor weight gain and failure to thrive. Steatorrhea occurs in patients who continue to take in a normal-fat diet. Malabsorption of fat is associated with severe malabsorption of fat-soluble vitamins. Without vitamin E administration in a water-soluble form, patients suffer progressive degeneration of the central nervous system. Another abnormality, probably caused by the lack of carotene and vitamin A, is decreased visual acuity and night blindness. Blood smears from many affected patients show spiny projections called acanthocytes (i.e., "thorn" or "spur" cells) on 50% to 70% of erythrocytes. Prothrombin time may also be prolonged as a result of vitamin K malabsorption
Mech: The exact underlying biochemical defect involves an abnormality in the synthesis or secretion of the lipoproteins containing Apo B. The result is severe malabsorption of fat due to failure of chylomicron formation and the restricted delivery of endogenously synthesized cholesterol to peripheral tissue via the LDL pathway.