
Immunology
Immune System
Evaluation of Immune Function
Primary Immunodeficiency Disorders
Autoimmunity and rheumatologic disease
HLA - see Transfusion
Immune System
Innate Immune System
Cells and proteins protect body from danger, intruders
Cells: Neutrophils, Monocytes/macrophages, Dendritic cells, NK cells
Proteins:
– Cytokines, e.g. IL-1, 6, TNF
– Complement? Lyses cells, attracts mediators
– C-Reactive Protein: Activates complement, opsonin
– Haptoglobin: Transport protein, binds hemoglobin
– Alpha-1 antitrypsin: Reduces inflammation
How are foreign/danger molecules recognized?
– Pathogen associated molecular patterns (PAMP) or Damage associated molecular patterns (DAMP)
– Recognized by pattern recognition receptors (PRR)
• Toll Like Receptors: Cell surface proteins, similar
to a “Toll” protein in Drosophila. Recognize bacterial motifs
• NOD(nucleotide oligomerization domain)-like receptors present in the cytoplasm
B cells
Originate and mature in bone marrow in stepwise process, where they undergo rearrangement of immunoglobulin gene
B= Bursa of Fabrecious: B cell development organ in
birds
Immunoglobulin
4 polypeptide genes, including 2 heavy chains and 2 light chains, bound by disulfide bonds
- light chains have a variable and a constant region
- heavy chains have 1 variable and 3-4 constants
The terminal constant region can insert into the membrane of B cells or, if free in serum, is called the Fc portion
- Fc can bind Fc receptors (FcR) on the surfaces of phagocytic cells
- mast cells bear an Fce receptor that renders them capable of binding IgE
- the variable regions of a light and heavy chain combine to form the ag binding site, the paratope
Immunoglobulin genes
Light Chain Genes
Variable and Joining (V and J) Regions
– 76 Kappa V Genes; 5 Kappa J Genes
• Separate V and J genes for kappa and lambda
- during B cell development, these genes are rearranged
• One V will combine with one J, then this VJ construct will be coupled to the C gene
- Light chain genes on cr 2 (kappa) and 22 (lambda)
heavy chain genes (gamma, alpha, mu, delta, and sigma) are on cr 14
Heavy Chain Genes
• V, D, and J regions
• 100-200 V genes; 6 J Genes
• App 30 D(iversity) genes
• V-D-J joining occurs
• Complete VDJ gene combines with the C gene
-- begins c variable region rearrangement of V&J (light chain) or V, D, and J (heavy chain) sequences
- then rearranged variable region sequence is joined to a light chain or heavy chain constant region gene sequence
- the latter is always initially a mu heavy chain gene, resulting in a completed gene that encodes an IgM protein with some particular epitope specificity
Mature B cells coexpress surface IgM and IgD
- once stimulated by ag in the presence of Th cell the mature B cell proliferate and each progeny B cell rearranges its DNA yet again, rejoining their prefacricated variable genes with a different heavy chain gene (isotype switch)
- T cell interactions and cytokines stimulate isotype
switching.
• Isotype switching occurs by Ch gene rearrangement to a “downstream” gene
The variable end of the ab is itself an epitope
- called the idiotope (idiotype)
- anti-idiotope (idiotype) abs can be formed against it
5 Ig classes, based on the heavy chain isotype:
1) IgG (IgG1, IgG2, IgG3, IgG4)
- monomers in the blood (2 binding sites)
- avg serum conc 120 mg/dL
- activates classic pathway
- 4 subclasses, IgG4 lowest concentration
2) IgA (IgA1, IgA2)
- dimers in the blood (4 binding sites)
- avg serum conc 30 mg/dL
- activates alternate pathway
- 2 subclasses, IgA2 lowest concentration
3) IgM
- pentamers in the blood (10 binding sites)
- avg serum conc 15 mg/dL
- activates classic pathway
4) IgD
- bound to B cells in the blood
- avg serum conc 0.3 mg/dL
- does not activate complement pathway
5) IgE
- bound to mast cells in the blood
- avg serum conc 0.0005 mg/dL
- does not activate complement pathway
There are also 2 light chain isotypes (kappa and lambda)
T cells
Undergo stepwise maturation in the thymus
- under normal conditions, T lymph outnumber B lymphs 2:1 and represent 65% of circulating lymphs
- T cell receptor (TCR) genes (alpha, beta, gamma, and delta) are present on cr 7 and rearrange in fashion similar to Igs
- T cells differentiate into several types that are separable on the basis of cell surface ags: T helper (Th) cells have CD4, T suppressor (Ts) and T cytotoxic (Tc ) cells have CD8; and CD4:CD8 ratio usually is ~2:1
T cell receptor (TCR)
- analogous to Ig but unlike Ig can only respond to epitopes presented in conjunction c MHC/HLA molecules
- CD4+ (Th) cells must have the ag presented in conjunction c class II MHC molecules (class I MHC restricted)
- CD8+ (Tc) cells must have the ag presented in conjunction c class I MHC molecules (class I restriced)
CD4 T Cell Subclasses
• Th1 Cells; produce IL-2, 12 and IFN: Promote cell
mediated immunity, responses to intracellular pathogens
• Th2 Cells; produce IL 4, 5, and 10: Promote humoral immunity, allergic responses, humoral autoimmunity
• Th0 Cells: Produce IL2, 4, and IFN
• Th17 Cells: Produce IL 17, activated by IL-23, also important in Autoimmune bowel diseases, and Response to gut bacteria
Memory Aid: Principal role of T cells is CMI. Thus,
Th1 help CMI, Th2 help Humoral Immunity (a
secondary T cell function)
T Regulator Cells
• CD4, CD25 (IL-2 receptor) positive
• suppress autoimmune responses
• May be involved in controlling anti-graft responses
• The elusive “suppressor cell”?
2 classes of TCR
1) most (>95%) T lymphs have TCR-ab
2) TCR-gd found in greatest number in mucosal surfaces and skin
TCR expressed in noncovalent assoc c CD3 molecule
NK cells
Represent ~10% of peripheral blood lymphs and have neither TCR nor IG. TCR and Ig genes are in the germline (nonrearranged) state. NK cells lack surface CD3 and express CD16, CD56, and CD57. CD16 is the receptor for the Fc portion of gamma heavy chains (FC-gamma R)
- through binding of opsonized cells c this receptor, they mediate ag dependent cellular cytotoxicitiy (ADCC); combat viral infx and tumor cells
- secrete IFN-gamma and are morphologically recognizable in the PB as large granular lymphocytes (LGL)
Antigen Presenting Cells (APCs)
Include monocytes, macrophages, histiocytes, and organ specific cell types such as Kuppfer cells of hepatic sinusoids, Hoffbauer cells in placental villi, interdigitating reticulum cells (IRC) of the interfollicular portions of lymph nodes, dendritic reticulum cells (DRC) of germinal centers and Langerhans cells in epidermis and lung
- have phagocytic properties and certain cell ags: MHC class II ags, CD68 (KP-1) lysozyme. Akk exceot the monocyte macrophage cells ahve S100 and CD1a
- secrete IL-1
Granulocytes
Neutrophils
Attracted by cytokines, esp IL-8 and upon arrival are stimulated to release granules into surroundings
- also capable of limited phagocytic activity
Basophils and mast cells
Have Fce receptors on surface and able to bind IgE
- if antigen (allergen) is present, the surface IgE can become crosslinked, which activates degranulation
Eosinophils
Stimulated by a subset of CD4+ T cells called Th2 cells that secrete IL-4 (stimulate production of IgE) and IL-5 (attract eosinophilic infiltration)
- degranulation can result in formation of Charcot-Leyden crystals (CLC); CLC protein has lysophospholipase activity
Complement
Series of over 30 proteins that work with antibody to effect lysis of target cells
– Involved in the inflammatory response
3 methods of activation
1) Classical
2) Alternative
3) Mannose Binding
• Rarely is only 1 pathway involved, but 1 pathway may predominate
Effects
The attachment of the complement protein C3b to foreign substance (opsonization) leads to phagocytosis
- the attachment of the complex of complement proteins C5-9 (membrane attack complex) leads directly to the lysis of membrane bound structures
- complement proteins C3a and C5a (anaphylatoxins) promote the release of histamine from basophils
Pathways
Classical pathway - activated by IgM and certain subclasses of IgG (IgG1-3)
- the Fc portion of Ig interacts c C1q to initiate this process
-- activation sequence: C1, 4, 2, 3, 5, 6, 7, 8, 9
- activated C1 catalyzes the assoc of C4 and C2 to make C4b2a (the C3 convertase of the classic pathway)
- C4b2a is capable of converting the C3 to C3b and C3a. The now abundantly available C3b assoc c C42a to make C42a3b (the C5 convertase of the classic pathway) causing deposition of the terminal complement components on the target surface
Alternative pathway - Properdin Factor B/Zymogen induced C3 Cleavage
- Once C3 is cleaved, the rest of the pathway will continue
- Fluid phase C3 convertase continues this pathway
- activated by bacterial cell walls, venoms, endotoxin, or complexed IgA
- initiation of this pathway is through deposition of C3b onto any of these substances
- C3b is being produced at low levels at all times in peripheral blood as a result of a "tick over" phenomenon but is quickly degraded, if not bound, y factor I
- bound C3b assoc c factor B to make C3bBb
- factor D cleaves the factor B to form the surface bound convertase (the C3 convertase of the alternate pathway) which is further stabilized y properidin
- C3bBb is capable of converting C3 to C3b and C3a, thereby amplifying the reaction
- the now abundantly abailable C3b assoc c C3bBb to make C3bBb3b, which is stabilized by properdin, sometimes written C3b(2) BbP (the C5 convertase of the alternative pathway)
Mannan binding lectin pathway (MBL) - uses mannose binding lectin (MBL) or mannose binding protein to bind onto the surfaces of microbes
- once the proteins bind, they undergo a conformational change allowing them to assoc c 3 MBL assoc serine proteases, MASP1, MASP-2 and MASP-3 to activate the complement cascade
- after activation, MASP-2 cleaves C4 and C2 to generate C4b2a, the C3 convertase, thereby activating C3
Basically, Mannose Binding Lectin Associated Proteases (MASP) 1 and 2 can cleave C4
• starts the classical pathway from C4 on
• assoc with Salmonella, Listeria, Neisseria, Candida
and Cryptococcal inf
The final result of the alternative, classical and MBL pathways is allowing for assembly of the membrane attack complex by producing C5 convertase (a molecule capable of converting C5 and C5b and C5a)
- once C5b is formed, it quickly complexes on the surface of adjacent cells c C6-9 to form C5b6789 (the membrane attack complex or MAC)
Human Leukocyte Antigens (HLAs)
- see Transfusion for more details
HLA proteins are encoded by genes located on the major histocompatibility complex (MHC) on cr 6p
HLA genes are categorized into classes:
Class I genes
- distributed among HLA-A, HLA-B, and HLA-C
- each locus has multiple possible alleles, termed HLA-A1, HLA-A2, etc
- these genes encode the HLA class I ags, which are heterodimers composed of 1 heavy chain and 1 light chain
- the heavy chain is a transmembrane polypeptide composed of 3 alpha domains
- it is noncovalently assoc c a light chain that is a single molecule of a2 microglobulin
- somatic cells that express foreign ag on their surface in conjunction c class I MHC molecules invite destruction from Tc (CD8+) cells
- class I ags are found on the surfaces of most nucleated cells
Class II genes
Distributed among HLA-DR, HLA-DP, and HLA-DQ
- for each locus there are several possible alleles, termed eg HLA-DR3, HLA-Dw2
- these genes encode HLA class II ags
-- each consist of 2 polypeptide chains, a and B each c 2 domains similar to the Ig light chains in addition to a transmembrane domain
- APCs present ag to Th (CD4+) cells in assoc c class II MHC ags
- HLA class II ags expressed on B lymphs, monos, macrophages, dendritic cells and activated T lymphs
Class III genes
- largely encode non-HLA ags, most of which still have something to do c immunity
- include genes for complement proteins, the NOTCH4 gene, the tumor necrosis factor (TNF) genes and others
- also embedded in the MHC region are the genes for gereditary hemochromatosis and 21-hydroxylase
Each MHC complex is closely linked and inherited
- each parental cr can be throught of as a haplotype
- thus the chance that 2 siblings are HLA identical is essentially 1/4
- the formula to calc the probability of having at least 1 HLA matched sibling c "N" siblings is 1-(0.75)^N
- threfore, the chance of having an HLA-identical sibling hoes up c the number of siblings: c 1 sibling the chance is 25%, c 2 is 45%, 3 is nearly 60%
MHC Deficiencies
- use to be called "Bare Lymphocyte Syndrome"
• Combined, Can be class I or class II deficiency
• Lack of either class I (rarer; vasculitis) or class II antigens (failure to thrive, infections) on the lymphocyte surface.
• Bone marrow transplantation may be helpful
• Autosomal recessive disorder
– Class 1 deficiency- TAP1, TAP2 genes
– Class II deficiency- RFXAP, RFXANK genes










Evaluation of Immune Function
Screening tests
History and physical exam
Primary immunodeficiencies disproportionately affect males (majority are X-linked)
- Ig or B-cell defects present c recurring bacterial infx of the upper and lower respiratory tract, ultimately resulting in bronchiectasis, or recalcitrant intestinal infx, eg c Giardia intestinalis
- T-cell defects result in susceptibility to viral and fundal opportunistic infections. Mucocutaneous candidiasis is an indication of defective T cell function
- phagocytic defects are assoc c Staphylococcus spp and other catalase positive organisms
- terminal complement deficiencies present c severe infx due to encapsulated orgs such as Streptococcus pneumoniae or Neisseria meningitides
- lymph nodes may be small or undetectable in some of the B cell defects, alternatively, nodes may be significantly enlarged in common variable immunodeficiency or chronic granulomatous disease
- petechiae and easy bruisability are seen in Wiskott-Aldrich syndrome
Global tests of immune system
Cell counts
Absolute lymphopenia is more common in T cell defects, since they comprise the majority of circulating peripheral lymphocytes
- absolute neutropenia may suggest inherited (constitutional) or acquired (autoimmune, drug) neutropenia
- TBCpenia may suggest such things as Wiskott-Aldrich syndrome
Radiographs
Radiologic imaging may disclose characteristic bony abnormalities
Specific testing of B cell function
Specific antibody response, eg to vaccine
Abs raised to protein ags require orchestration of T and B cell function, whereas B cells are capable of autonomous production of ab to carbohydrate ag
- a poor response to a protein ag such as tetanus or diptheroid toxoid could indicate either a B cell or a T cell defect, whereas poor reaction to carbohydrate ags (pneumococcal or meningococcal vaccine) is indicative of a B cell defect
Ig levels
- the MC isolated isotype deficiency is IgA deficiency
- IgG subclass deficiency is less frequent
- total IgE may be elevated in a subset of conditions (eg hyper IgE [Job] syndrome)
RAST (RadioAllergoSorbent Test)
- an allergen specific IgE measurement that can be useful to evaluate allergy, principally for inhaled allergens
Specific testing of T cell function
By flow cytometry, the proportion of T cell subsets can be determined
- a test of delayed type hypersensitivity (DTH) is the usual screening test for T cell function. A tuberculin skin test is classic
- proliferation assays can be performed by exposing T cells to mitogens such as phytohemagglutinin or concanavalin A. Proliferation is measured by the uptake of radioactive DNA precursors (tritiated thymidine)
Testing NK cell function
NK cell function can be assessed by chromium release assays
- a colorimetric substrate to detect granzyme B, a serine protease present in the granules of NK cells and cytotoxic T cells can be used to assess NK cell function
Testing neutrophil function
Absolute neutrophil cound and peripheral smear morphology
- examination of chemotaxis, phagocytosis or oxidative burst functions with various assays
- cells capable of a normal oxidative burst will reduce yellow NBT to a purple blue formazan (F) precipitate and are said to be F+. Normal indiciduals will have nearly 100% F+ cells. An abnormal result, with perhaps <10% F+ cells, is expected in chronic granulomatous disease (CGD), in which deficiency of NADPH oxidase presents the oxidative burst
- flow cytometry can detect intracellular oxidation of dihydrohodamine 123. When these products are oxidized they will fluoresce and are objectively evaluated by the instrumentation, an improvement over the subjective interpretation of the NBT dye test
- myeloperoxidase staining can disclose myeloperoxidase deficiency, an autosomal recessive trait which produces at most mild immunodeficiency
Testing complement
CH50
The CH50 test is a functional assay of the classical complement pathway, in which total hemolytic activity is measured
- the endpoint of the assay is the dilution of the pts serum causing 50% lysis of the Ig coated sheep red cells
- the result is expressed as the reciprocal of this dilution
AH50 tests the MAC (C5-9), more or less
Ags assays are undertaken for quantitation of specific complement components
Decreased levels of C3 refelct either primary C3 deficiency or activation of the alternate complement pathway
- levels of C4 or C1q are typically used to look at the classical pathway
HLA testing
Used primarily in:
- pretransplantation compatilibility testing
- plt refractoriness
- paternity/forensic identity testing
- evaluation of HLA linked autoimmune disorders
The complement dependent cytotoxicity (CDC) assay is the gold standard for:
- detecting HLA ags
- detecting HLA abs
- performing HLA crossmatching
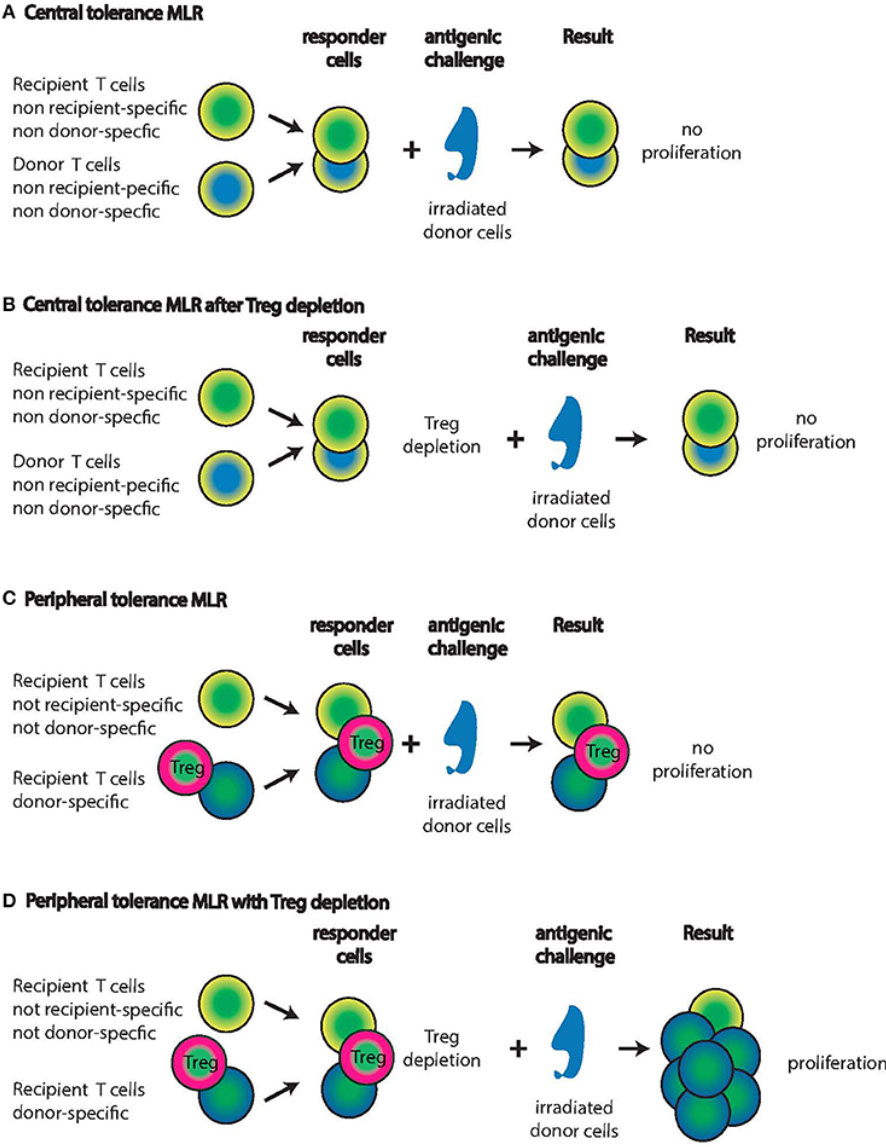
Mixed lymphocyte culture (MLC)
- assay capable of detecting HLA class II (HLA-D) differences among potential donor and recipient
- this test detects mainly HLA class II differences, which are of paramount importance, second only to ABO compatilibility, in organ transplantation
Cross reactive ag groups (CREGs)
- human antisera are capable of binding to more than 1 HLA allelic product (serologic crossreactivity)
- serologic crossreactivity bwt alleles of HLA-A and HLA-B loci are used to cluster molecules within the same crossreactive ag groups (CREGs)
Public antigens
- HLA ags contain common amino acid sequences that are present in many different HLA ags but demonstrate common reactivities
- these "public" ags are present in the less variable regions of the HLA molecule
- for example, HLA-Bw4 and HLA-Bw6 are 2 public ags that are present in almost all HLA-B molecules c only a few exception. If a pt lacks one of these public ags and becomes immunized during preg or transfusion, it appears as multiple discrete HLA abs. These abs can impact pt responses to plt transfusion or their ability to receive a compatible solid organ transplant
DNA assays
- have become widespread in HLA typing
- PCR testing has the advantage of eliminating many of the biologic uncertainties of the "serologic" techniques described above. Furthermore, it can resolve HLA types with much greater specifity than the serologic techniques
Transplantation testing
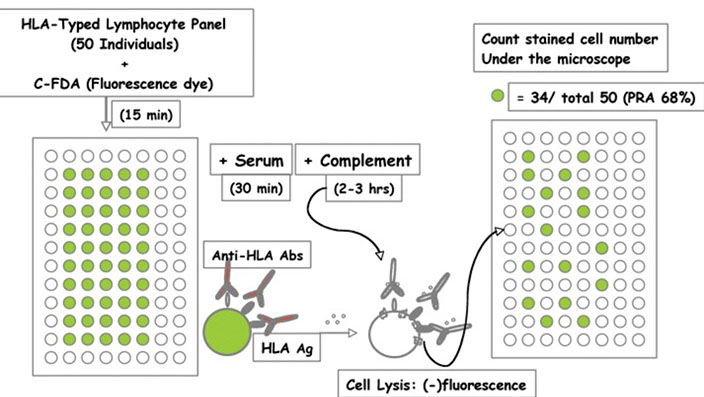
Panel reactive ab (PRA) tests
- used to monitor pts who are waiting for a transplant
-- these pts may be sensitized to HLA ags either through transfusion of donor blood products, preg or previous organ transplantation
- PRA assesses the proportion of the donor population that the pts HLA abs will react against, giving some guidance to the rate of compatibility of an unrelated donor organ. For example if the pt has a PRA of 80%, this indicated the pt is highly sensitized and has about a 20% chance of compatibility with an unrelated donor organ that becomes available
HLA matching for transplantation
- usually involves matching at least 3 loci: HLA-A, -B and -DR
- since each person has 2 alleles (one on each 6p) for each locus, there are 6 possible alleles
The CDC crossmatch
- can detect preexisting HLA allaabs in the serum of the potential recipient that have specificity for HLA ags in the potential donor
- these preformed HLA abs are the mediators of so called hyperacute rejection, in which the transplanted organ is rapidly destroyed by circulating abs
- positive results of a CDC crossmatch bwt donor and recipient indicate an incompatibility, and the organ should not be transplanted into the pt
The flow cytometric crossmatch
- a more sens test than the CDC assay
- allows for detection of lower titers for both complement dependent and complement independent recipient abs to the donor graft
Luminex technology
- has been correlated c flow crossmatches and CDC testing
- Luminex values:
-- > 5000 MFI correlates c a positive T cell and B cell flow crossmatch
- > 10,000 MFI correlates c a positive CDC , indicating hyperacute rejection is likely if the organ is transplanted to the pt
For renal transplantation, organs should be:
- ABO compatible
- HLA-A, -B, -DR matched (6 of 6 ideally)
- crossmatch compatible
For transplantation of heart, lung, and liver
- such stringency is not required
- ABO compatibility is the main determinant for donor selection; HLA typing can be performed but is not a requirement
HLA typing resolution requirements for allogenic progenitor cell transplants
- more stringent than for renal transplants
- it is optimal to match the donor and recipient alleles at the HLA-A, B, C and DRB1 loci
Transplant rejection
Hyperacute rejection
- occurs within hours of transplantation
- mediated by preformed high titer abs to ABO or HLA ags expressed by donor graft endothelium
- Ig is deposited along vessel walls, inducing complement mediated vascular injury
- the result is the formation of fibrin and plt thrombi, causing ischemic necrosis
Acute cellular rejection
- evolves over days to weeks after transplant
- mediated by T cells
- recipient T cells recognize foreign HLA ags and stimulate a powerful cellular cytotoxic response
- histologically, a lymphocytic infiltrate is seen within graft epithelium (tubulitis) and endothelium (endothelitis) along with interstitial edema
Acute humoral rejection
- present within days to weeks after transplantation
- mediated by ab
- like hyperacute rejection, the endothelium is the primary target, with resulting injury taking one of 2 forms
- first, there may be a pattern similar to hyperacute rejection, with fibrinoid necrosis and neutrophilic infiltration of vessel walls and resulting graft infarct
- second there may be a more insidious vascular subendothelial intimal thickening with more protracted graft ischemia
- pts who are HLA alloimmunized as a result of pregnancy or transfusion are at significantly higher risk. In either case, immunohistochemistry demonstrates deposition of C4d in vessel walls
Chronic rejection
Mediated by a combination of lymphocytes and antibodies over months to years, leading to eventual graft failure
- histologic findings include interstitial fibrosis, arteriosclerosis of vessels and complement deposits in peritubular vessels
Graft vs host disease (GVHD)
3 requisites for development of GVHD:
- immunocompetent donor T cells
- immunosuppressed recipient
- ag differences vwt donor and recipient
Acute GVHD
- occurs within the first 100 days after transplant, c most cases presenting within the first 30 days
- 3 main targets of acute GVHD:
1) Skin - initially characterized by an erythematous, pruritic rash, histologically demonstrating characteristic erythema, multiforme-like apoptosis that is most pronounces at the bases of rete ridges
2) Intestinal tract - presents c diarrhea. Histologically, colon bx show ectatic crypts c attenuated enterocytes, crypt abscesses and striking apoptosis within the crypt epithelium
3) Hepatobiliary tract - liver involvement presents c jaundice, histologically characterized by mononclear portal inflammatory infiltrates c endothelialitis, ductitis, and ductopenia
Chronic GVHD
Occurs after the first 100 days
- affects the skin, hepatobiliary tract, intestinal tract, and the mucosa of the mouth, vagina, eye and respiratory tract
- these pts experience extensive cutaneous sclerosis that resembles progressive systemic sclerosis. They may experience esophageal strictures, bronchiolitis obliterans, scarring ocular lesions and chronic liver damage
- this is different from transfusion associated GVHD (TA-GVHD) in which the main target of the attack is the bone marrow
- the rate of death in TA-GVHD is over 90% due to aplasic anemia and can be prevented by irradiating lymphocyte containing blood products in immunocompromised pts
RAST


CH50

Mixed Lymphocyte Reactions (MLR)
Panel Reactive Antibody (PRA) Assay for Cytotoxic Anti-HLA Antibodies


Pathophysiology of GVHD

Primary Immunodeficiency Disorders
B cell defects
Bruton (X-linked) agammaglobulinemia, aka Bruton's Thymidine Kinase (BTK) Deficiency
Recurrent bacterial infxs, particularly related to encapsulated bacteria that is predominantly 2/2 antibody deficiency, beginning ~ 6 mo age (after maternal ab wanes)
- normal immunity against most viral and fungal pathogens; increased susceptibility to polio, hepatitis, and enteroviruses
- high incidence of lymphoid neoplasms and autoimmune diseases
- serum immunoglobulin levels (IgG, IgA, and IgM) are markedly reduced, marked dec CD19 B cells
- lymph nodes and tonsils are rudimentary
- the responsible gene, BTK, is found on the Xq22 chromosome. It encodes a tyrosine kinase called Btk (Bruton tyrosine kinase)
--there is an AR form of the s yndrome that is related to defects in the genes encoding the u heavy chain gene, CD79A and CD179B
Tx: IV Ig
Transient Hypogammaglobulinemia Infancy with normal numbers of B cells
• Presents approximately 5-6 mo, similarly to Bruton's, though these pts have normal CD19 B cells
• IgG and IgA low, IgM may be normal to low
• Recurrent upper respiratory infections
• CD19 positive B cells ARE present
• Hypogammaglobulinemia may last up to 2 years.
• Cause is undefined
Common variable immunodeficiency (CVID)
- typical onset is in 2nd or 3rd decade
- recurrent upper and lower respiratory tract infections, intestinal bacterial overgrowth and Giardia intestinalis infection; bronchiectasis common
- low serum Ig (IgG, -M, -A) and variable T cell deficiency
May be a B or T cell defect
– Th1 imbalance with elevated IL12 and gamma interferon
– Autosomal recessive, ICOS defect
Selective IgA deficiency
Recurrent respiratory and gastrointestinal bacterial infections
- high incidence of autoimmunity
- at risk for anaphylaxis due to transfusion of IgA containing blood products
- more common inherited Ig disease (1 in 700 ppl)
Predominately Antibody Deficiency
- MC primary immune deficiency. Occurs in 1 in 700
- IgA levels <5 mg/dl, IgG and IgM are usually normal.
- concomitant IgG2 deficiency (IgA with IgG subclass
deficiency) maSy beel eprcesteinvt.e IgA, 2
• Recurrent respiratory infections, allergies, autoimmune diseases and GI tract diseases may be present, but many IgA deficient people have no symptoms
• Pts at risk for an allergic, non-hemolytic transfusion reaction if antibodies to IgA are produced.
This may give a serum sickness type of reaction to a
whole blood or plasma transfusion, or an Arthus (IgG) or anaphylactic (IgE) type of reaction to a subsequent
transfusion.
Isolated IgG Subclass Deficiency
• Predominately Antibody Deficiency
• May present at any age
• Recurrent respiratory infections, IgG2 (and possibly IgG4) deficiencies result in encapsulated organism infection. IgG1 and IgG3 deficiencies result in infections with toxin producing organisms.
Dx: monitor immunoglobulin response to protein (tetanus, IgG1 and IgG3) and/or polysaccharide (pneumococcus, IgG2 and IgG4) vaccines
CD40 Ligand Deficiency
• Combined Immunodeficiency
• Presents in the first year of life
• IgM levels are normal to elevated (100-1000 mg/dl)
• IgG, IgA and IgE levels are low.
• CD4 cell inCcreDase4 m0a yL oicgcuarnd Def, 2
• Defect is in the gene (Xq26 ) coding for the T cell protein, CD154 (CD40 Ligand) molecule.
– CD154 binds to CD40 initiating class switching in B
cells
– This defect prevents T cells from inducing B cells to
undergo a class switch. Thus the B cells produce IgM, without switching to another isotype.
• Autosomal recessive form does exist, involving the
TNFRSF5 gene
Hyper IgE syndrome (Job syndrome)
Recurrent staphylococcal infx, coarse facial features, and elevated IgE
- eosinophilia and eczema
- AD and AR forms
Increased IgE
Wiskott-Aldrich syndrome
Job syndrome
Nezelof syndrome
Decreased IgE
Bruton agammaglobulinemia
Ataxia-telangiectasia
T cell defects
DiGeorge syndrome
- aka congenital thymic aplasia or hypoplasia
Failure of the 3rd and 4th pharyngeal pouches to develop, hypoplastic thymus and parathyroid, anomalies of the great vessels, typical facies (hypertelorism, low set ears, mandibular hypoplasia), bifid uvula, and a higher than usual incidence of esophageal atresia
- may present with neonatal hypocalcemic tetany
- partial DiGeorge syndrome more common than complete DiGeorge syndrome; some cases present as CHARGE sequence (coloboma, heart defect, choanal atresia, retardation of developemnt, genital hypoplasia, and ear anomalies)
- T cell defect results in susceptibility to opportunistic fungi, viruses, and Pneumocystis jiroveci
- can have some "pre" T cells expressing CD1 and CD7
- inc risk of transfusion assoc GVHD
- LN paracortical areas are depleted, and poorly developed periarteriolar lymphatic sheaths (PALS) are found in the spleen
- microdeletions involving cr 22q11.2, Tbx1 mutation
- occur sporadically; some cases assoc c in utero exposure to Accutane
Tx: monitor heart dz and hypocalcemia
- prophylaxis for opportunistic dz can help
- thymic transplants have been used
Severe combined immunodeficiency (SCID)
Dec/absent T cell function, low/undetectable Ig, and thymic dysplasia
- not really 1 disease, but several diseases
- T, B and LGL are low or absent. Deficiencies are listed as T, B, and NK, followed by a + or – to indicated whether or not those cells are present
- several forms of SCID exist, usually presenting shortly after birth to 6 mo of age
- life threatening immunodeficiency and inc risk for TA-GVHD
- bacterial, fungal, viral and protozoan infx
- variety of genetic defects
IL-2 receptor gamma chain mutation (Xq13); also affects receptors for IL-4, 7, 9 (4, 7, and 9 tell bone marrow to make more lymphs), 15 and 21. Phenotype is TB+ NK+or-
• Adenosine deaminase (ADA) deficiency (20q13, autosomal recessive). Phenotype is T- B-NK-
• JAK3 Deficiency (T-B+)
Tx: aggressive anti-microbial; bone marrow
transplant.
Hyper IgM syndrome (X linked immunodeficiency with hyper IgM)
- impaired isotype class switching with low levels of IgG, IgA, IgE and inc IgM
- clinical picture similar to Bruton agammaglobulinemia
- X linked inherited disorder caused by defect in gene that encodes CD154, the T cell ligand for the B cell receptor CD40
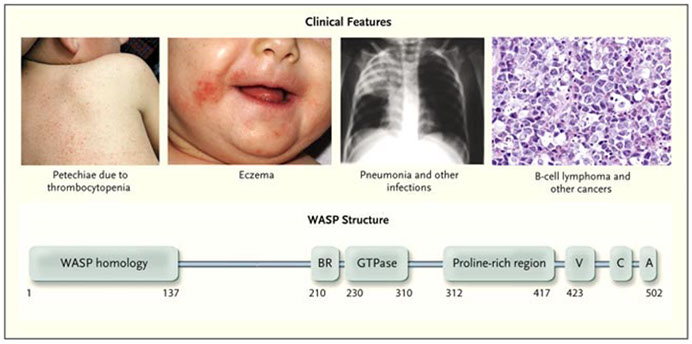
Wiskott-Aldrich syndrome (WAS)
X linked inherited disorder that presents with the triad of eczema, thrombocytopenia, and immunodeficiency
*** TIE *** Thrombocytopenia, Immunodeficiency, Eczema ***
- presents in the first year of life
- susceptibility to infx by Pneumococci and other encapsulated bacteria, Pneumocystic jirobeci and herpesvirus
- the WAS gene is located on the X chromosome (Xp11.2), encodes Wiskott-Aldrich syndrome protein (WASP)
- IgE and possibly IgA are elevated, IgM is usually low
- T cells are present but with a reduced response to mitogens
Ataxia telangiectasia (Louis-Bar syndrome)
Cerebellar ataxia, oculocutaneous telangiectasia, recurrent sinopulmonary infx, and a high incidence of malignancies
- combined T cell and B cell defect
- lab abnormalities include deficient IgA along c very high serum AFP and CEA
- AR, caused by mutations in ATM gene on 11q22.3
Chronic mucocutaneous candidiasis
Selective defect in T cell immunity to candidal infx
- assoc c endocrinopathies and a variety of autoimmune disorders
- mutations in autoimmune regulator gene (AIRE), chromosome 21q22.3
Duncan disease (X linked lymphoproliferative disease)
Fulminant and often fatal immune response to Epstein-Barr virus (EBV) infection
- often have an immune system defect like CVID
- inverted CD4:CD8 ratio and hypogammaglobulinemia common
- the underlyinh genetic abnormality is found in the SH2D1A gene, Xq25, which codes for an SH2 domain on a signal transducing protein called SLAM assoc protein (SAP)
Neutrophil / phagocytic defects
Chronic granulomatous disease (CGD)
Defective intracellular oxidative killing of ingested organisms, most commonly resulting from deficiency of NADPH oxidase
- chronic suppurative infections with catalase positive bacteria, esp Staphylococci, Enterobacter, and Aspergillus; streptococcal infections not common
- extensive granuloma formation at sites of infection
- the screening test is the nitroblue tetrazolium (NBT) test; oxidative burst can also be measured by chemiluminescence or flow cytometry
- affected leukocytes are deficient in C3b receptors, Red cells ofter bear the McLeod phenotype (absence of Kell ags, Kx)
Chediak-Higashi syndrome
AR condition that presents as neutropenia, recurrent infx, TBCpenia, and oculocutaneous albinism
- defective degranulation
- granulocytes, lymnphocytes, and monocytes show giant cytoplasmic granules
- Mutation in the lysosomal trafficking regulator (CHS1/LYST) gene at 1q42.1-2
May-Hegglin anomaly
AD condition manifesting as Dohle-like bodies in granulocytes and monocytes, variably-sized plts, and TBCpenia
- Dohle-like bodies abolished by ribonuclease
Alder-Reilly anomaly
Mainly a morphologic finding
- large azurophilic granules resembling toxic granulation in all WBCs
- assoc c mucopolysaccharidoses
Jordan anomaly
Characterized by fat vacuoles in leukocyts
Complement deficiencies
Deficiency of classical pathway components (C1q, C2, C4, C5) is primarily manifested by autoimmune phenomena such as lupus
- deficiency of C3 and C3 leads to recurrent infx c Gram+ encapsulated organisms
Deficiency of membrane attack complex components (C5-9) also referred to as terminal complement components, leads to recurrent serious system infx, esp 2/2 esp Neisseria meningitidis and Neisseria gonorrhea
C1 esterase inhibitor (C1 INH) deficiency manifesting as hereditary angioedema (HAE) can be tested with C1INH
- urinary histamine levels and serum C1 levels are elevated during attacks, while serum CH50, C4, and C2 are decreased. Between attacks, C4 is always low, while C2 levels are normal
Tx: in the past, acute episodes were treated c infusions of donor plasma; however recombinant C1 inhibitor concentrate became available in 2008 and is now the first line therapy for these patients
- other therapies include a kallikrein inhibitor (ecallantide) and a bradykinin B2 receptor (icatibant) antagonist; androgenic agents (danazol, oxandrolone and methyltestosterone) are used as prophylaxis

Bruton Agammaglobulinemia
Four genetic defects in cell surface receptors on lymph in CVID pts. The figure highlights their cellular expression pattern, interacting molecules, and major biological functions

Clinical Anomalies in CVID

CD40L Deficiency (and others)





SCID
Wiskott-Aldrich syndrome

Autoimmunity and Rheumatologic Disorders
Pathophysiology
Tolerance acquisition occurs during fetal development in the thymus
- autoimmunity occurs when there is a breakdown in self tolerance
- MHC dz assoc (see chart)
Autoimmune dz's are most prevalent in women of reproductive age
- exceptions worth noting are ankylosing spondylitis, which affects males more often than females, and Sjogren syndrome that affects postmenopausal females
Triggering agents include both microorganisms and medications
- Hepatitis B virus (HBV) infection can trigger polyarteritis nodosum (PAN); infection may provoke ankylosing spondylitis
- the drug aldomet can lead to autoimmune hemolytic anemia; penicillamine is linked to systemic vasculitis
- procainamide, hydralazine and isoniazid are assoc c drug induced lupus
Laboratory testing, general
- common findings include anemia of chronic dz and elevation of acute phase reactants
- platelets may be elevated (acute phase reactant)
- may have either leukocytosis or leukopenia, and lupus in particular is assoc c lymphopenia
- the aPTT may be prolonged if there is a lupus anticoagulant
- nonspecific inflam markers include CRP, ESR, ferritin, and fibrinogen; ESR often used to monitor dz activity
- complement levels can serve as markers of dz activity in immune complex deposition disorders such as lupus
- synovial fluid analysis may disclose mononuclear cell predominant (RA), neutrophil predominant (septic, gout), histiocyte predominant (PVNS), or pauci-inflam (DJD) patterns
- Lupus erythematosus (LE) cells are neutrophils with ingested lymphocyte nuclei
-- the lymphocyte nucleus should be devoid of chromatin detail and stain pink in Wright stained preparations
- may be seen in pleural effusions, joint effusions, or bone marrow aspirates and provide strong evidence for SLE
-- the "tart" cell mimics the LE cell but retains chromatin detail, and chromatin stains blue purple
- Histologically, RA and RA-like conditions demonstrate marked papillary synovial hyperplasia and usually dense infiltrate of lymphs and plasma cells, often c nodular lymphoid aggregates within which there may be germinal centers
Autoantibody detection by immunofluorescence
Direct immunofluorescence (DIF)
- involves incubating cryostat sections of patient tissue with fluorescein labeled antihuman globulin (AHG)
- positive DIF tests confirm the in vivo presence of bound autoabs in the pts tissues
Indirect immunofluorescence (IIF)
- involves incubating pt serum c cells / tissue known to contain specific ags, then adding fluorescein labeled anti-human globulin (AHG)
- positive IIF tests confirm the presence of circulating autoabs
Antinuclear antibodies (ANA)
- may be detected by IIF, incubating pt serum c HEp-2 cells; pattern of fluorescence both in the mitotically active and resting cells is noted, and all positives are serially diluted to determine titer
- negative ANA virtually excludes systemic lupus erythematosus (SLE)
- positive ANA is not specific for SLE
-- 20% of normal adults have an ANA titer of 1:40, and up to 5% have a positive ANA with a titer of 1:160; false positives inc c age
- non-SLE conditions assoc c a positive ANA include other autoimnune diseases (Sjogren syndrome, scleroderma, rheumatoid arthritis), multiple sclerosis, infx, malignancy and fibromyalgia
-- when a cutoff titer of 1:40 is used, the specificity is ~80%; when 1:160 is used, specificity is ~95%
- to help confirm the dx of SLE, additional testing for anti-double stranded (ds) DNA, anti Smith (Sm) and antiphospholipid abs often undertaken; however these tests have lower sens for SLE
- high titer anti-RNP (>1:10,000) abs are characteristic of mixed connective tissue disease (MCTD), particularly if unaccompanied by other ANAs. Anti-RNP is commonly seen in SLE, but titers are usually modest
- anti SS-A / Ro and anti SS-B / La abs c neg dsDNA and Sm are compatible with Sjogren syndrome
- anti-Scl-70 (antitopoisomerase 1) is seen exclusively in progressive systemic sclerosis (PSS)
- anticentromere abs are strongly suggestive of CREST syndrome and are occasionally seen in PSS and Raynaud syndrome
- drug induced lupus is usually negative for dsDNA, Sm and RNP but is characterized by positive ANA and antihistone abs directed at the H2A-H2B dimer complex
- the terms "extractable nuclear ags" and signal recognition particles" refer to subgroups of ANAs
-- extractable nuclear ags include Smith (Sm), ribonucleoprotein (RNP), SSA (Ro) and SSB (La)
- signal recognition particles (SRPs) include Jo-1 and PM/Scl. SRPs are stronglt assoc c inflam myositis
- anti dsDNA in the ketoplasma of the organism Cribidia lucilae
Antibodies to cytoplasmic constituents
Sceening for abs to cytoplasmic constituents performed by IIF using rat tissue as substrate, ie rat liver, kidney and stomach that are "sandwiched" together
- examined for pattern of fluorescence and all positives are titered
- in certain instances this approach has been replaced c ELISA
Antimitochondial ab (AMA)
- demonstrates reactivity c the cytoplasm of gastric parietal cells, renal tubular cells, and hepatocytes
- directed against a mitochondial ag called M2
- assoc c primary biliary cirrhosis (PBC)
Anti smooth muscle ab (ASMA) reacts c the smooth muscle of the gastric tissue and renal arterioles. Anti liver kidney microsomal (anti LKM) ab reacts c the cytoplasm of hepatocytes and renal tubular epithelial cells. Both ASMA and anti LKM assoc c autoimmune hepatitis
Antineutrophil cytoplasmic antibody (ANCA)
Refers to a pattern of reactivity obtained when pt sera are incubated c alcohol fixed neutrophils
- testing for ANAs is performed on all positived to exclude a false positive ANCA due to the presence of ANAs
- cytoplasmic ANCA (cANCA) appears as a cytoplasmic granular pattern c perinuclear accentuation; cANCA has antiproteinase 3 (PR3) specificity. cANCA is highly specific for Wegener
- Perinuclear ANCA (pANCA) is characterized by predominantly perinuclear immunofluorescence and has antimyeloperoxidase (MPO) activity. pANCA is prsent in microscopic polyangiitis (MPA), polyarteritis nodosum (PAN) primary sclerosing cholangitis (PSC) and ulcerative colitis (UC)
Antithyroid antibodies by immunofluorescence
- detected by incubating serum c cryostat sections of thyroid and fluorescein labeled AHG
- fluorescence that highlights follicular epithelial cell cytoplasm is consistent with antithyroid peroxidase (antimicrosomal) antibody. fluorescence that highlights follicular content on methanol fixed tissue is consistent with antithyroglobulin
Automated techniques such as EIA are supplanting fluorescence based testing in many labs
Rheumatoid factor (RF)
An autoab that binds to the Fc portion of the IgG molecule
- a marker of rheymatoid arthritis (RA)
- can be found in health adults and a wide range of non-RA disease conditions, most commonly other rheumatologic dzs, chronic infx, viral infx, and hematolymphoid neoplasms
Anticitrullinated protein (anti CCP) abs have greater specificity and sensitivity for RA than RF
- levels may correlate with dz activity
Angiotensin convertin enzyme (ACE)
An extremely useful test in the evaluation of pts c suspected sarcoidosis, in which it is nearly always elevated when the dz is active
- other causes of elevated ACE are primary biliary cirrhosis (PBC), Gaucher dz, and leprosy
Autoimmune diseases
Systemic lupus erythematosus (SLE)
Female to male ratio ~8-9:1; most pts diagnosed bwt the ages of 15 and 45 years; greater prevalence in African Americans
- presentation is variable from pt to pt
- most have rash (90%), arthralgia (75%), neurologic sx (65%), serositis (60%), hematologic (anemia and TBCpenia in 60%), Raynaud phenomenon (45%), vasculitis (40%), glomerulonephritis (30%) and endocarditis (15%)
America College of Rheumatology criteria for dx are intended for research purposes; 4 of 11 must be fulfilled for the dx of SLE
Negative ANA essentially excluded lupus, other autoabs may be present
Autoabs in SLE and frequency
ANA (>99%), anti dsDNA (40-90%), anti single strand DNA (70%), anti Ro (SSA) [24-60%], anti La (SSB) [9-35%], anti Sm (15-35%), anti RNP (35%), anti cardiolipin and/or LAC (25%), antiribosomal P (10%)
Drug induced lupus
Generally follows a benign course
- assoc c procainamide, hydralazine, quinidine, isoniazid, and interferon-a
- antihistone abs are the hallmark of drug induced lupus; usually directed against the H2A-H2B dimer complex protein of the histone
Rheumatoid arthritis
MCC of chronic inflammatory arthritis; extraarticular manifestations include interstitial lung dz, pleuritis, endocarditis, and systemic vasculitis
- women affected about twice as often as men, initial onset of symptoms between the ages of 30 and 50 years; inc incidence in pts c HLA DR4
- C-reactive protein and erythrocyte sedimentation rate are elevated; RA is a common cause of anemia of chronic dz, and platelets are often mildly elevated as an acute phase reactant
- positive rheumatoid factor (RF) in most pts, but up to 30% of pts initially have a negative RF
- anticyclic cirullinated peptide (anti CCP) antibody is significantly more specific than RF
- synovial fluid analysis is notable for elevated white count c preponderance of neutrophils and low glucose (c negative cultures and no crystals)
Seronegative spondyloarthropathies
Grp of disorders that have sx in common c rheumatoid arthritis but are RF-, including ankylosing spondylitis, reactive arthritis (previously Reiter syndrome), and psoriatic arthritis
- all are more common in males and in persons c the class I human leukocyte ag allele HLA B27
- Reactive arthritis is provoked by antecedent mucosal infx, typically urethritis (Chlamydia) or gastroenteritis (shigella, Salmonella, Yersinia, and Campylobacter)
Celiac disease
Tends to cluster within families, and there is a strong assoc c HLA-DQ2 and HLA-DQ8
- often assoc c other autoimmune conditions, esp type I diabetes mellitus and autoiimune liver dz
- increased incidence in pts c IgA deficiencym cystic fibrosis, Turner syndrome, and Down syndrome
- histologically, the duodenum demonstrates villous atrophy, intraepithelial lymphocytosis and crypt hyperplasia
- prolonged inflammation may result in malnutrition with or without iron folate deficiency
- prolonged inflammation assoc c lymphoma (enteropathy assoc T cell lymphoma)
- extraintestinal manifestations include arthritis and dermatitis herpetiformis
- seologic markers include antigliadin, antiendomysium, antireticulin and anti tissue transglutaminase (tTG) abs. IgA anti tTG is considered the best first line test; some recommend excluding IgA deficiency before interpreting this test
Progressive systemic sclerosis (scleroderma)
Manifestations range from Raynaud phenomenon to scleroderma to widespread dz involving the GI tract, heart, lungs, and kidneys; leading causes of death include pulmonary hypertension and renal crisis
- limited cutaneous PSS (CREST syndrome) usually manifests as scleroderma limited to the distal extremities, without cardiac, renal or pulmonary involvement; manifestations described by the CREST syndrome: Calcinosis, Raynaud, esophageal sclerosis, sclerodactyly, and telangiectasia
Labs: autoabs found in PSS include ANA , anticentromere, anti Scl-70 (antitopoisomerase), anti U1 RNP (antifibrillarin), anti RNA polymerase 3, anti Th/To, and anti Pm/Scl; anti Th/To and anti Pm-Scl are responsible for antinucleolar reactivity identified as specific for PSSl anticentromere, anti Th/To, anti U1-RNP and anti Pm-Scl are assoc c limited cytaneous (CREST) SS
IgG-related sclerosing disease
- causes fibroinflam lesions such as sclerosing mediastinitis, idiopathic retroperitoneal fibrosis, Reidel thyroiditis, sclerosing cholangitis, orbital pseudotumor, and sclerosing pancreatitis
- biopsies display a lymphoplasmacytic infiltrate, rich in IgG4 producing plasma cells
- elevation of serum IgG4 is found in most (>90%) cases; IgG4 is normally the minor IgG subclass, representing ~5% of total IgG
Autoimmune hepatitis (AIH)
- lab findings include hypergammaglobulinemia (polyclonal IgG) and abnormal liver function tests, including prolonged Pt/INR and raised AST/ALT
- in AIH type I, ANA, ASMA, and antisoluble liver protein (anti SLP) may be present
- AIH type II is characterized by anti liver kidney microsomal ab (LKM1) an ab against CYP 2D6
Primary Biliary Cirrhosis (PBC)
- liver function test abnormalities have a cholestatic pattern with prominence of alkaline phosphatase and GGT; also assoc c PSS and CREST
- 80-90% with IgM polyclonality
- antimitochondrial abs (AMA), subtype M2, are present in >95% of cases; IgG anti sp100 and IgG anti gp210 are also highly specific for PBC
- pts often have hypercholesterolemia and polyclonal hyperimmunoglobulin M
Primary sclerosing cholangitis (PSC)
More common in males and in pts c UC
- pANCA may be positive, but AMA is negative
- dx made by cholangiography
Sjogren syndrome (SS)
Characterized by dryness of the eyes, mouth and other mucous membranes
-- ~1% incidence, 9/10 are female
- can present with dry mouth and eyes (SICCA), keratoconjuunctivitis, possibly secondary to lymphocyte infiltration
- 1/2 are primary, 1/2 secondary (to RA)
- may be primary or assoc c other systemic autoimmune dzs (secondary SS), particularly RA, PBS, SLE or systemic sclerosis
- anti SS-A/Ro and anto SS-B/La abs are characteristic but are not specific
- histologic dx depends upon minor salivary gland bx showing >1 inflam focus / 4 mm^2, with a focus defined as an aggregate of at least 50 mononuclear inflammatory cells
- pts at inc risk of lymphoid malignancy
Vasculitis - see Vascular Pathology
The vasculitis syndromes can be roughly divided into those c fairly precise serologic markers and those without; fortunately, those without serologic markers have highly characteristic clinical features
Mixed Connective Tissue Disease
~4/5 female; Presents at any age with Raynaud’s phenomena and swollen digits, Pulmonary disease, Polyarthralgias; Lungs (~85% of patients) with Pulmonary hypertension, dyspnea
– Heart- pericarditis
– Renal disease due to IC
– Severe neurological symptoms
Overlap syndrome, similar to SLE
– Anti-RNP (without anti-Sm)
– Myositis with progressive weakness
– Thickened, taught skin
– Polyarthralgias
Hypersensitivity Reactions
Type I hypersensitivity (immediate type hypersensitivity)
Result of ag binding to IgE on the surfaces of mast cells (bound by the FceR); IgE induced crosslinking of FceR results in degranulation, with the release of histamine, heparin, serotonin and arachidonate
- neither anaphylactoid reactions nor hereditary angioedema are type I hypersensitivity reactions
- anaphylaxis is a type I hypersensitivity reaction mediated by IgE. Serum (or plasma) is elevated in anaphylaxis (chronically elevated in systemic mastocytosis). Plasma histamine levels peak more quickly than tryptase, at ~10 minutes after the onset of anaphylaxis. Levels return to normal within an hour. The urinary histamine, however, may be elevated for 24 hours
Type II hypersensitivity (antibody mediated cellular cytotoxicity)
- produced by ab-ag binding that leads to tissue damage mediated by opsonization
- examples include Goodpasture syndrome, myasthenia gravis, autoimmune hemolytic anemia and erythroblastosis fetalis
Type III hypersensitivity
- mediated by the formation of immune complexes as a result of ab-ag interaction with activation of complement
- examples include Arthus rxn, SLE, HSP, serum sickness, PSGN, MGN and the artrus rxn
Type IV hypersensitivity (delayed type hypersensitivity)
- mediated by CD4 T cells reacting to ag, usually c granuloma formation (macrophages)
- example is the tuberculin skin test, poison ivy

Cytoplasmic antibodies
Antimitochondrial antibody (AMA)
Fluorescent staining pattern - cytoplasm of gastric parietal cells, renal tubular cells and hepatocytes
Disease assoc - primary biliary cirrhosis
Anti smooth muscle antibody (ASMA)
Fluorescent staining pattern - gastric smooth muscle and renal parenchymal arteries
Disease assoc - autoimmune hepatitis
Antiparietal antibody (APA)
Fluorescent staining pattern - gastric parietal cells
Disease assoc - pernicious anemia, atrophic gastritis
Anti liver kidney microsomal (LKM)
Fluorescent staining pattern - cytoplasm of hepatocytes and renal tubular cells
Disease assoc - Autoimmune hepatitis
p-ANCA
Fluorescent staining pattern - perinuclear staining of neutrophils
Disease assoc - primary sclerosing cholangitis, ulcerative colitis, microscopic polyangiitis, polyarteritis nodosum (PAN)
c-ANCA
Fluorescent staining pattern - cytoplasmic staining of neutrophils
Disease assoc - Wegner granulomatosis, microscopic polyangiitis
Clinically useful abs
Anti dsDNA
Also called anti native DNA, has high specificity for SLE. The substrate, Crithidia lucilae, is a flagellate with a giant mitochondrion that contains double stranded DNA concentrated in an area celled the kinetoplast. Abs to ssDNA do not react c C lucilae
Anti Jo 1
Also called anti tRNA synthetase
Polymyositis (PM), esp PM with pulmonary fibrosis
Antihistone
High specificity for drug induced systemic lupus; assoc drugs include hydralazine, procainamide, isoniazid, dilantin, methyldopa and penicillin
Extractable nuclear antigens
A number of ags, the so-called extractable nuclear ags (ENAs), are present in the extract of calf thymus. ENAs typically give a speckled pattern on fluorescent ANA testing. ENAs include Smith, ribonucleoprotein (RNP), SS-A (Ro) and SS-B (La)
Anti Smith (Sm)
Virtually diagnostic of SLE
Anti RNP
Mixed connective tissue diseae (MCTD), esp if ANA is negative
Anti nucleolar
Scleroderma (PSS)
Anti SS-B/ SS-A
SS-A (Ro) is present in 70% of pts with Sjogren syndrome and 30% of pts c SLE. SS-B (La) is present in 50% of pts c Sjogren and 15% of pts c SLE. Anti SS-A/SS-B are highly prevalent in neonatal lupus. Pts who are ANA+, SS-A+ but SS-B- are very likely to have lupus nephritis
Antimitochondrial (AMA)
AMA is detected in 85% of pts c orimary biliary cirrhosis (PBC). AMA is reactive c the cytoplasm of parietal cells of the stomach and renal tubular cells in the mouse stomach / kidney substrate
Anti smooth muscle (ASMA)
Lupoid (autoimmune) hepatitis is characterized by titers of >1:80. ASMA are specifically directed at F-actin. ASMA are detected on the mouse stomach / kidney substrate
Anti LKM
Autoimmune hepatitis
Antimicrosomal
High specificity (90%) and sensitivity (95%) for Hashimoto disease, although up to 50% of pts c Graces dz will have abs
Antiendomysial ab
Endomysin is present in the reticular investment of muscle fibers; is an immunofluorescent assay used on monkey esophagus. Antiendomysial abs are IgA abs, They are highly sensitive and specific for celiac sprue and dermatitis herpetiformis. Ab titers respond to a gluten-free diet
Antigliadin
Gliadin is a protein derived from gluten; both IgG and IgA anti-gliadin abs seen in Celiac sprue
- IgG is usually more sens, while IgA is more spec
Anti-striated muscle antibody
Cross reacts with thymic epithelium; seen with myasthenia gravis
Anti-acetylcholine receptor ab
Blocks neuro-muscular transmission, seen in myasthenia gravis
- AchR blocking ab in 1/2 of pts
Anti tissue transglutaminase (tTG)
- enzyme that can deamidate gliadin, the target is the anti-endomysial ab; ELISA and multiplex tests for anti-TTG abs are objective and have inc sens and spec compared to the anti-endomysial IFA test
- IgA tTG may be the best test for Celiac sprue
Anti GBM
Goodpasture syndrome; the epitope is the M2 subunit of type IV collagen (GBM = Glomerular Basement Membrane)
Anti IF
Polymyositis / dermatomyositis (IF = Intermediate Filament)
Anti Scl-70
Anti Scl-70 ab is an antitopoisomerase Ig seen in 20% of pts c scleroderma
Anti PM1
Overlap syndrome c overlapping feature of scleroderma and dermatomyositis
c-ANCA
High specificity for Wegener syndrome; cANCA is antiproteinase 3 (PR3)
p-ANCA
Microscopic polyaniitis, eosinophilic granulomatosus with polyangitis (Churg-Strauss)
Antithyroglobulin
Hashimoto dz
Thyroid stimulating ab
Also called LATS, it is present in 90% of individuals c Graves dz
Anti RNA polymerase 3
Scleroderma (PSS)
Antitopoisemerase (Scl-70)
Scleroderma (PSS)
Antiribosomal P
PM, DM
Type 1 diabetes (DM1)
Glutamic acid decarboxylase (GAD65), tyrosine phosphatase (IA-2), insulin (IAA), and Zinc transporter 8 (ZnT8A)
Hu antigen
Marker found in all cells of nervous system, also seen in small cell ca
- marker assoc c polyneuropathy
Rheumatoid factor (RF)
Rheumatoid arthritis
Anti CCP
Rheumatoid arthritis

Rheumatoid Arthritis criteria



Sjogren syndrome

MCTD criteria

References
1.