
Hematology
Intro to blood
Hematopoiesis
Maturation sequences (Wbc, Rbc, Plt)
RBC Morphology and the CBC
RBC molecular structure & metabolism
Hemoglobinopathies
Thalassemia
Immune hemolytic disorders
Defects in marrow production
WBC kinetics
Thrombocytosis
Intro to Blood
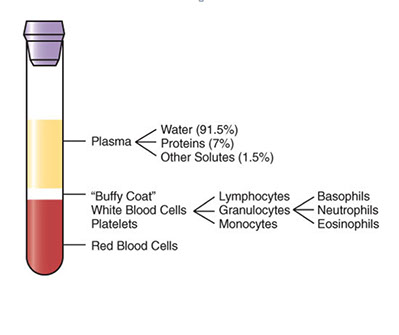
Blood composition
Plasma - liquid protein matrix c nutrients, proteins, hormones, electrolytes, metabolic biproducts
Cellular component: RBC, WBC and plts (red, white and blue!)
- buffy coat contains WBCs and plts; and is zone bwt plasma and RBCs after whole anticoagulated blood centrifuged
- for CBC blood should be drawn in dipotassium ethylenediamine tetra-acetic acid (K2EDTA)
Serum = liquid component of blood after allowed to clot; lacks coagulation factors and platelets
Plasma = natural state in body, clotting factors not activated and no plts consumed, the liquid component of anticoagulated blood
Blood transports O2, has regulatory proteins, nutrients, and removes waste products
- also serves as defense c coagulation and hemostasis and inflam and ab production c wbcs
Hematology is the study of cellular elements of the blood and can determine px, confirm dx, monitor dz progression
- must eval cellular elements of blood c smear, where drop of blood smeared and stained c Romanowsky polychrome dye or Wright-Giemsa stains (Leishman and May-Grunwald stains seldom used)
- Wright stain has Methylene blue (azures A, B, C, stains acid [basophilic] structures shades of blue / purple), eosin Y (halogenated fluorecent dye that stains basic [acidophilic] structures shades of pink, orange or red), absolute methanol (fixative, diluent for powedered stain), and phosphate buffer (maintains pH 6.4-6.7, thus controlling color development)
Normal RBCs
- carry O2, size of 6-8 microns, no nucleus, red cytoplasm c central pallor 1/3 of cell, no granules
- life span: 120 days
WBCs
*** Never Let Monkeys Eat Bananas!!! Neuts, Lymphs, Monos, Eos, Basos ***
Neutrophils: reference range 38-79%, most numerous WBC in normal pb, which fights infx, is 12-16 microns (2-3x RBCs), nucleus segmented into 2-5 lobes (aka Polymorphonuclear cells [PMNs] or stabs), low NC ratio, coarse chromatin c cytoplasmic granules (tiny pink tan)
Lymphocytes - ref range 12-51% or 0.6-3.8 x 10^9/L, produces abs and mediate immunity, 6-15 micron size, round-oval nucleus thats usually eccentric and can be indented, moderate to high NC ratio, dark clumped chromatin +/- nucleoli, blue cytoplasm that can be scant to abundant, usually absent granules, though NK lymphs have red granules
Monocyte - ref range 0-10% or <0.6 x 10^9; fights infx and cleans up cellular debris; 14-20 microns, indented partially lobed nucleus, moderate NC ratio, fine lacy chromatin +/- nucleoli, lots of blue-gray "dishwater" cytoplasm that can have pseudopods, can have tiny barely visible granules or ground-glass cytoplasm
Eosinophils - ref range 0-8% or <0.6 x 10^9/L;
*** NAACP: Neoplasia, Addisons, Allergy, Collagen vascular dz, Parasites ***
- size: 13 microns, low NC ratio, coarse , clumped chromatin that's a little lighter than neutros, no nucleoli, large red-orange uniform granules
Basophil - ref range 0-1%, or <0.1 x 10^9; assoc c allergy, also neoplasia / dysplasia sometimes, 12 microns, nucleus bilobed but usually obscured by dark blue polymorphic granules, same nuclear chromatin as neutro
Platelets
- involved in hemostasis, usually less than 4 microns in size, no nucleus, pink-purple cytoplsm c tiny uniform granules
- live 7-10 days
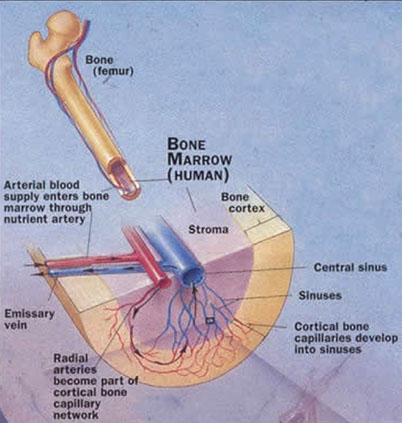
Hematopoiesis (HP)
The production of blood cells, mostly in the BM, called medullary hematopoiesis (thymus and LN also play role in differentiation and maturation of lymphs); extramedullary hematopoisis is outside the BM, mostly in the spleen and liver, and occurs in stressful situations, with resulting H/S-megaly
- all blood cells come frm a Hematopoietic stem cell, which are capable of self-renewal and multi-lineage differentiation
In a bone marrow biopsy (bmbx), must assess Myeloid:Erythroid (M:E) ratio which is normally 3:1 or bwt 2-4:1; and assess morphology in all cell lineages
- must assess cellularity (100% newborn, 50% adult, 30-40% if >60 yo, or can use 100-age to estimate); can be hypo-, normo- or hyper-cellular
- mkc's assessed on core for relative abundance and morphology
- iron is judged on the aspirate and clot c prussian blue stain on a scale of 4 (0= absent, 1 trace, 2-3 normal, 4 increased)
- also do flow, cytogenetics, FISH and molecular studies
- contra-indications for bmbx are lidocaine allergy, hemophilia, obesity
Primary lymphoid organs = BM and thymus
- site of lymphocyte birth and maturation by a process that does not involve antigens; have no follicles and make mature naive lymphocytes
- BM has HP and stromal (non-hp, provides nutrients) compartments
-- HP compartment shows Erythroblastic islands clustered central macrophage (macrophages secrete cytokines [CKs] to reg erythroblast maturation); Granulocytic clusters (granulocytes divided into proliferating and storage pools); and Megakaryocytes (mkc's; adjacent to sinus endothelium)
- the BM stroma is all tissue not directly involved in HP, providing microenvt and infrastructure allowing HP to occur; cells of the stroma secrete CKs and GFs for HP, stroma includes: osteoblasts, osteoclasts, fibroblasts, adipocytes and endothelial cells
Secondary lymphoid organ (LNs, spleen, MALT, tonsils) activate naive lymphocytes with antigen and have follicles
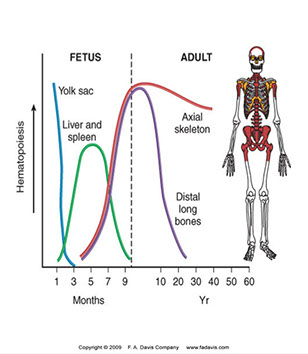
HP begins in fetal development early on
- in first trimester (1-3 months GA), occurs in the yolk sac
- in second trimester (4-6 mo GA) in liver and spleen
- in third tri (6-9 mo GA) BM takes over
- throughout life (after childhood) HP recedes from limb bones towards axial skeleton and ends on long bones, which is why bmbx done on post iliac crest of adults and upper end of tibia in kiddos
Cytokines (CKs) are soluble glycoproteins secreted by cells for cell-to-cell communication
- hematopoietic growth factors are types of cytokines targeting hematopoietic cells
- blast's lineage cannot be determined by morphology alone!!! need help of CD markers
Growth factors reg HP c promoting precursor cell prolif, reg diff and maturation, programmed cell death (apoptosis)
- binding sites are CD markers
-- monoclonal ab's discovered in 70s, WHO began studying specificity of monoclonal ab's, where each ag given a "Cluster of Differentiation" (CD) designation, determined by cell type and maturation phase of target cells; >300 CDs recognized today
- types of growth factors:
Lineage-restricted growth factors - act on single cell line
- EPO, TPO, GM/G/M-CSF
Synergistic growth factors - act on multiple cell lines; ie interleukins


Maturation Sequences
HSC = Hp Stem Cell (not committed to any cell line)
HPC = Hematopoietic Progenitor Cell (committed to either myelo or lymphoid cell lines)
- CLP = Common Lymphoid Progenitor
- CMP = Common Myeloid Progenitor (aka Colony forming Units [CFUs]; CFU-Granulocytic-Erythroid-Mkc-Monocytic [CFU-GEMM]
- CD markers help identify lineage and maturation stage
- as cells mature, cell size dec (except promyelocytes), nuclear size dec (chromatin condenses and nucleoli less visible); nuclear color changes (more purple, less red); cytoplasmic changes (more to less blue); inc granularity
-- nomenclature: _blast --> Pro_cyte --> _cyte --> Meta_cyte
--- ie Myeloblast, Promyelocyte, Myelocyte, Metamyelocyte, band, granulocyte (N,E,B)
WBC maturation sequences
Myeloid lineage
Myeloblast
- blue cytoplasm (from lots of ribosomes), prominent golgi (from inc synth and transport, pushed nucleus eccentrically, mitochondria clustered around golgi causes juxtanuclear clearing), nucleus c 1-5 prominent nucleoli
- IL3 and G-CSF causes myelocytic differentiation
Basos and eos have same maturation sequence as netros...
Promyelocyte
CD Markers- CD33 & CD38; Size: 15-24 µm (larger than myeloblast!); N:C ratio- 5:1- 3:1 (lower than blast); Nucleus eccentric (from golgi) c reddish, lacy chromatin that begins to condense around perimeter
- 1-2 nucleoli may be seen, less visible; Cytoplasm c juxtanuclear clearing and contains many reddish primary granules that increase as becomes less of a blast; capable of mitosis
Myelocyte
CD Markers-CD45RA; Size: 10-18 µm (close to a mature cell); N:C ratio 2:1; Nucleus may be eccentric (less golgi) but is more clumped, less red more blue; No / rare nucleoli (less mitoses); Cytoplasm clear to bluish-pink (still some RNA activity)c 20 granules appear; Specific to N, B, E
- Last stage capable of mitosis
Metamyelocyte
CD Markers- CD15; Size- 10-18 µm; N:C Ratio: 1:1; Nucleus indented which is < 1/2 diameter of rounded nucleus; Chromatin bluish-purple and clumped; Can see both chromatin & parachromatin; Cytoplasm clear, pink; Specific granules present
Band neutrophils
CD Marker: CD15; Size: 10-16 µm; N:C ratio: 1:1-1:2; Nucleus indented and > 1/2 diameter; Thick bridge of chromatin separating lobes; Chromatin & parachromatin are visible; Nuclear chromatin dark, condensed, bluish-purple; Cytoplasm pink-clear; Specific granules present
Segmented neutrophil
CD Marker: CD15; Size: 10-16 µm; N:C ratio: 1:3; Nucleus c 3-5 lobes separated by thin filament; Dark, clumped; chromatin & parachromatin visible; Cytoplasm c specific granules present
Monocytic series
Monoblast
Size- 15-25 µm; N:C ratio- 7:1-4:1; May look like myeloblast
- Can only distinguish using CD Markers (CD64- Fc receptor; CD14- co-receptor for LPS [“Monocyte differentiation antigen”]
- Nucleus open, lacy reddish-purple and may be folded c 1-3 prominent nucleoli; Cytoplasm dark blue, agranular c intensely stained perimeter
Promonocyte
Intermediate between Monoblast & Monocyte; Size: 14-20 µm;
IHC: (+) CD13, CD33, CD11b, CD14, CD15, HLA-DR
-- NOT possible to distinguish a promonocyte and monocyte based on IHC
Nucleus starts to indent, but chromatin is immature; may have “cerebreform folding”, fewer nucleoli visible; N/C ratio: 4:1-2:1; Cytoplasm less blue than Monoblast but more blue than a monocyte; N:C ratio higher than Monocyte
- Vacuoles may be present in cytoplasm and may contain rare azurophilic granules
Monocyte
CD Markers-CD14; Size: 20-25 µm; N:C ratio: 1:2-1:8; Monocytes are immature macrophages and may differentiate into dentritic cells
- Nucleus lobulated/indented; no visible nucleoli
- Cytoplasm gray c many non-specific granules give “ground glass” appearance to cytoplasm; occasional azurophilic granules and may have vacuoles
Macrophage
Size: 25-80 µm; CD Marker: CD68; most mature cell in this lineage
- May be fixed in tissue or recruited to site of inflammation
- Macrophages never seen in peripheral blood!
Dendritic cell
Size: 15-20 µm; CD Markers- depends on stage of development, and function: CD11c, CD21, CD123, CD35, CD141, CD303
- Function as Antigen Presenting Cells (APCs)
- in PB Dendritic cells look like monocytes, can only be distinguished by CD markers (?)
- Dendritic cells may also arise from the lymphocytic line
Lymphocytic Series
All lymphs descend from Common Lymphoid Progenitor (CLP) in BM
- B cells differentiate and mature in bone marrow
- T cells migrate to the thymus to mature and differentiate
- NK (Natural Killer) cell differentiation is poorly understood
- May take place in the bone marrow, thymus, or lymph nodes
- CD Markers are the best way to identify stages of maturation and differentiation.
Lymphoblast
CD Markers- many; Size-10-20 µm; N:C ratio- 7:1; Cytoplasm scant and very blue
- Nucleus red-purple, c smooth chromatin and 1-2 nucleoli
Prolymphocyte
CD Markers- many; Size-9-18 µm; N:C ratio- 5:1-3:1;
- Cytoplasm more visible, blue c juxtanuclear clearing
- Nucleus may be indented, chromatin becomes more condensed, blue-purple chromatin and reddish-purple parachromatin; 1-3 nucleoli
Mature Lymphocyte
Size: 7-15 µM; N:C ratio 4:1-2:1;
- Cytoplasm c active golgi; mitochondria near golgi causes juxtanuclear clearing; may see azurophilic granules in NK lymphs
- Nucleus eccentric and may be indented by golgi c condensed chromatin and “Wagon Wheel” appearance
Plasma Cell
CD Markers: CD27, CD138; size: 10-20 µm;
- Function: Ab production
- Cytoplasm royal blue cytoplasm c large golgi causing juxtanuclear clearing, causes nucleus to be very eccentric
- Nucleus very condensed c “Cartwheel” or “checkerboard” appearance and is eccentric
Plasma cell variants
Reed-Sternberg Cell
“Owl’s Eyes” appearance; present in nodular biopsies in Hodgkin’s Lymphoma, but may be present in other conditions
Strong negative predictive value- (if not present, most likely not Hodgkin’s Lymphoma)
- Caused by a defective B cell clone
CD15, CD30 positive
CD20, CD45 negative
Morula, Mott, & Grape Cells
Seen in cases of hyperglobulinemia (inc plasma cells in BM)
- Once thought to be all the same or the same as Russell bodies
- has identical appearance but different staining qualities
Flame Cell
Plasma cell with intensely red cytoplasm seen in IgA Myeloma, Waldenström’s Macroglobulinemia, African trypanosoma leptomeningitis
- Reddish tinge due to presence of IgA which contains large amounts of carbohydrate



Myeloblast


Promyelocyte
Promyelocyte
Myelocyte


Myelocyte
Metamyelocyte

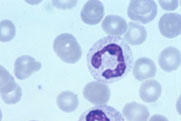
Bands
Segs
Monoblast


Promonocyte


Monocyte

Monos

Prolymphocyte
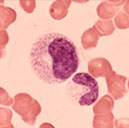
Plasma cell
Mature lymph
RS-cell


Flame cell
Morula/Mott cell

Erythrocytic Maturation Sequences
Erythropoiesis occurs in the BM
- Stem cells differentiate into Erythroid cells in response to erythropoietin stimulation (EPO produced in the kidneys, and regulated by a negative feedback loop involving oxygen)
- Adequate O2 delivery to the tissues is dependent upon adequate quality and quantity of RBC
- If the RBC mass dec, oxygen carrying capacity dec
BFU-E is the first to express erythropoietin receptors, in response to IL-3 stimulation
- High erythropoietin concentration stimulates expression of more erythropoietin receptors and differentiation into the CFU-E
- From the CFU-E stage on, differentiation is completely dependent upon erythropoietin
As erythroid cells mature, the following changes take place:
↓ Cell size; ↓ Nuclear size; Nuclear chromatin condenses, nucleoli disappear , nuclear color changes from red to more bluish purple and nucleus is eventually extruded
- Cytoplasm turns dark blue to pink/red
2 nomenclatures for RBC maturation:
1. Rubriblast - Prorubricyte - Rubricyte - Metarubricyte -Reticulocyte
Erythrocyte
2. Pronormo*blast - Basophilic normoblast - Polychromatophilic normoblast - Orthochromatic normoblast - Polychromatophilic Erythrocyte - Erythrocyte
* One nomenclature substitutes “normo” with “erythro”
Rubriblast (Pronormoblast)
14-24 μm; N:C ratio- 8:1 to 6:1; Cytoplasm c large golgi; Deep royal blue cytoplasm- “blue velvet” - 2/2 large amounts of RNA; Perinuclear halo; Mitochondria form circle around nucleus
- Nucleus reddish-blue, 1-2 large nucleoli; “ropy” coarse chromatin
Lavender parachromatin
Prorubricyte (Basophilic Normoblast)
12-17μm; N:C ratio- 6:1 to 4:1
- Cytoplasm lighter blue but still dark; Perinuclear halo less prominent or absent
- Iron stain yields siderotic granules
Nucleus purple, nucleoli not prominent; Clumps of chromatin
Rubricyte (Polychromatic normoblast)
10-15 μm (about the same size as a lymphocyte); N:C ratio- 4:1 to 2:1
- Cytoplasm more abundant and grayish-blue (due to hemoglobin production)
- Siderosomes present (iron stain)
- No perinuclear halo
- Nuclear chromatin more clumped, purple; Visible parachromatin, lavender; No nucleoli
*** rubricyte is the last stage capable of mitotic division
Metarubricyte (Orthochromic [acidophilic] normoblast)
8-12 μm; N:C ratio- 1:1 to 1:2
- Cytoplasm in moderate amount, pinkish gray
- Siderocytes may be present
- Nucleus eccentric, ready to be extruded and also pyknotic- compressed chromatin due to degeneration
Reticulocyte (Polychromatophilic erythrocyte)
7-10 μm; No nucleus
- Cytoplasm bluish-pink; May have basophilic stippling
-- Blue coloration & stippling caused by residual RNA
- May be visualized with supravital stain (Methylene blue)
Mature Erythrocyte
7-8 μm; Cytoplasm pink, no nucleus
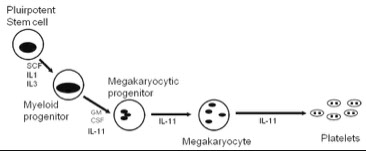
Megakaryocytic Maturation Sequence
Thrombopoietin Platelets are the only formed elements of blood which are not true cells; they are the cytoplasmic fragments of megakaryocytes
- Megakaryocytes are largest HP cell in BM
- Mkc growth stimulated by Thrombopoietin (Tpo)
- TPO produced in liver, to lesser degree in spleen (possibly BM and endothelial cells)
- Regulation of TPO availability is by a negative feedback loop
- TPO attaches to receptors on mkc and plts where it is destroyed
- The more platelets in the circulation, the more thrombopoietin is destroyed - when platelets are scarce, clearance of TPO is low, so concentrations increase
Definitions:
Endoreduplication - replication of the nuclear genome in the absence of cellular division, which leads to an elevated nuclear gene content and polyploidy.
Polyploidy- the condition of cells which have more than the normal two sets of chromosomes, each of which was inherited from one of two parents
Multipotential stem cells diff to mkc in response to TPO stimulation
- megakaryoblast begins nuclear division without cellular division- endoreduplication
- Endoreduplication (also called endomitosis) results in a large, multinucleate cell- polyploid nucleus
- A typical megakaryocyte progenitor undergoes three cell cycles without cellular division, resulting in a large cell with abundant cytoplasm and a multi-lobed nucleus
- Thousands of platelets arise from one megakaryocyte
- Unlike with other hematopoietic cells, megakaryocytes get larger as they mature
Megakaryoblast
20-40 μm; Smaller in pathologic conditions; N:C ratio 5:1-3:1
- Nucleus single, round or oval, eccentric
- Chromatin- red/purple, no clumps, 1-2 nucleoli
- Cytoplasm blue, moderate, may have prominent golgi, no granules
- The megakaryoblast Is able to leave the bone marrow and travel to extramedullary sites.
- Megakaryoblasts can divide
- IHC: (+) CD41 and 61, CD34, HLA-DR, CD9
-- neg CD13, CD33, and MPO
Promegakaryocyte
20-80 μm; N:C ratio 3:1- 1:1
- Nucleus may be single or double (2N or 4N)
- Cytoplasm blue and more abundant; fine, azurophilic granules may be seen arising from the golgi; psuedopods begin to form
Megakaryocyte
30-100 μm; N:C ratio 1:1-1:2
- Nucleus lobulated, 2 or more lobes; may be 8N to 64N; coarse, purple chromatin
- Cytoplasm pale blue and contains azurophilic granules
- Can see demarcation lines
- Platelets bud off at this stage
Other multinucleated cells in bone marrow
Osteoclasts
Megakaryocytes may be confused with osteoclasts in the BM
- Osteoclasts are multi-nucleate, however, the nuclei are separate
- mkc nuclei are attached
- CD markers are the best way to distinguish the two.


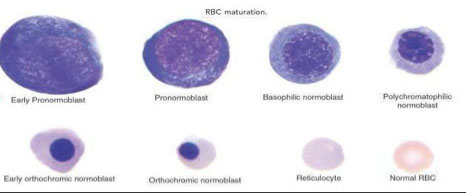
Proerythroblast

Basophilic erythroblast


Polychromatic erythroblast

Orthochromic erythroblast
Reticulocytes


Mature erythrocytes
megakaryoblast

promegakaryocyte


RBC Morphology and the CBC
Automated Hematology Instruments
Advantages of automation: Saves time; More standardized; Reduces identification errors (Especially if tubes are barcoded) ; Gives more information about cells than can be obtained using manual methods
Coulter Principle
Using a vacuum device, cells suspended in an electrolyte fluid are pulled through an aperture and into a tube.
An electrode is placed in the electrolyte solution, as well as inside the tube. Current is passed between the two electrodes.
Cells pass in single file through the small orifice, which causes a disruption in the flow of electricity.
This electrical pulse is sensed and recorded on an oscilloscope.
Each pulse represents a cell. Larger cells produce a large disruption, and thus a taller spike on the oscilloscope.
Data obtained is plotted on a number vs size histogram.
98% of all hematology instruments still based on the Coulter Principle.
Coincidence passage- algorithm which corrects for small cells that pass through at the same time and are counted as one.
Thresholds- electronically set size limits that exclude unwanted particles (below the threshold) and allow the selection of particles to be analyzed (above the threshold). Thresholds also distinguish between cell types (such as red cells and platelets) and sort cells within a population into subgroups (WBC types).
The following represent improvements made upon the original design and are used in modern day flow cytometry:
Hydrodynamic focusing- Diluted sample is injected into a stream of fast-moving sheath fluid which prevents mixing and assures that cells flow through the sensing zone in single file
Gating- sorting cells according to size and complexity of their nucleus.
VCS- Volume, Conductivity, and light Scatter is used to determine size and nuclear complexity to differentiate white cells.
Histograms and Scatterplots
A two-dimensional graphic representation of cell number versus one measured cell property (usually cell size). The number of particles (Y-axis) versus the size of particles (X-axis)
- Scatterplots - Three-dimensional graphic representations of two or more cell properties or characteristics plotted against each other (e.g.,number of cells vs cell size and internal cell structures such as granules or lobes).
Complete Blood Count
The Complete Blood Count is a collection of laboratory tests that evaluate the cellular components of the blood.
Frequently used as a screening procedure, it may also aid in diagnosis and treatment.
The CBC is nearly always done by automated instruments, except under extraordinary circumstances.
Cellular component:
WBC White Blood Cell Count- number of WBC/Liter
RBC Red Blood Cell Count- number of RBC/Liter
Plt Platelet Count- Number of platelets/Liter
Methods:
Aspiration: standardized amount of blood aspirated by instrument.
Dilution: blood is sent to two diluting chambers.
Sorting (RBCs and platelets*):
- RBC chamber, anucleate cells within specific size range counted. RBCs and platelets* are counted in this chamber.
- WBC chamber, RBCs lysed, and hemoglobin read
Sorting (WBCs): WBCs counted, then sorted according to size, the size and complexity of their nucleus, and the contents of the cytoplasm
Graphing: The data obtained from the counts is plotted on graphs.
Newer automated cell counters incorporate immunofluorescence into the identification of cells.
*Platelets may be counted in either the WBC or RBC chamber, depending on the instrument
Oxygen Carrying Capacity:
Hb Hemoglobin- concentration of Hemoglobin/dL
Method :
Measuring hemoglobin in both manual and automated methods is the cyanmethemoglobin method.
- WBCs are diluted in a reagent (Drabkin’s) that lyses red blood cells.
- Drabkin’s reagent converts all forms of hemoglobin except sulfhemoglobin to cyanmethemoglobin.
- reagent causes color change which is read by spectrophotometer.
- In most automated instruments, done in the WBC counting chamber.
- Cyanmethemoglobin Assay sources of error:
Elevated WBC count > 50 X 109/L WBC and/or nRBCs
Lyse-resistant hemoglobins; e.g., Hemoglobin S or C
Lipemia ; Bilirubinemia
Hct Hematocrit- packed red cell volume (total % of blood which is RBCs);
- In manual methods the hematocrit is measured directly, while in automated methods it is calculated.
Direct measurement
Whole blood is drawn into a capillary tube
The tube is spun at a standardized time and RPM
- percentage of red cells per total volume of blood in tube is read
In manual methods, the MCV is calculated from the spun Hct & the RBC count: MCV = HCT x 10 / RBC
Indirect measurement
In automated methods, MCV is measured directly, but the hematocrit must be calculated:
HCT=MCV x RBC/10
“Rule of three” used to check of the validity of your instrument results:
RBC x 3 = Hb +/- 0.5
Hb x 3 = Hct +/- 2.0%
RBC x 9 = Hct +/- 3%
It is used as a quality check of result you have already obtained; it must not be used to obtain a reportable result.
Troubleshoot specimen integrity if any of the above are out for clots, agglutination, hemolysis or lipemia.
If the above rules are out on several specimens in a row, troubleshoot your instrument.
Morphology:
MCV Mean Cell Volume- Size of Average RBC
- Relates to normocytosis, microcytosis or macrocytosis; a normal RBC should be about the size of the nucleus of a small lymphocyte
Normal size: 6-8 μ diameter
Normal MCV: 77.0 – 96.0 fL
Microcytic: < 75 fL ; indicate decreased output, as in iron deficiency anemia, thalassemia, and anemia of chronic disease
Macrocytic: >98 fL ; indicate either increased output (young cells), or a production defect such as megaloblastic anemia.
- Before the MCV could be measured directly by hematology instrumentation, it was calculated using the following formula:
MCV = HCT/RBC x 10
- The normal range for an MCV is 77-96 Femtoliters (fL)
MCH Mean Cell Hemoglobin- Average amount of Hb/RBC
MCH = HGB/RBC x 10
- The normal range for the MCH is 24.5-32 picograms (pg)
MCHC Mean Cell Hemoglobin Concentration- Average concentration of Hb/RBC; Relates to chromicity (hypo-, normo-, or hyperchromic)
- normal range MCHC = 30-35.
- Because an instrument measures the Hematocrit indirectly, it calculates the MCHC differently than in manual methods:
Manual MCHC = HGB/HCT x 100
Instrument MCHC = Hgb/(MCV/Rbc) x 10
- automated MCHC may be erroneous if cells agglutinated or sickled, because the MCV and RBC will be inaccurate
- Lipemic specimens also give high MCHC by interfering with the hemoglobin reading (If MCHC >37, specimen investigated for lipemia)
-- If lipemia is present, the plasma should be replaced with an equal volume of saline and the sample should be rerun.
-- If no lipemia is present, a blood smear should be evaluated for agglutination, sickling, clots, etc.
- hyperchromic stuff caused by dec membrane-to-vol ratio (spherocytes), while hypochromic stuff usually occus c microcytosis
RDW Red Cell Distribution Width (relates to anisocytosis on the blood film) is a measurement of the variation of red cell volume.
- It is the Coefficient of Variation of the MCV:
RDW = (Standard deviation of MCV ÷ MCV) x 100
- The RDW is increased when there is a wide variation in RBC size, and decreased when the cells are a uniform size:
Normal RDW 12.0-15.0%
Anisocytosis: > 17%; Low Abnormal: <10%
A high RDW indicates increased RBC output from the bone marrow
A low RDW suggests decreased RBC output from the bone marrow
MPV Mean Platelet Volume- Size of average platelet
Manual Differential
- done if instrument flags any abnormalities in automated differential.
- Manual differentials use Romanowsky stains to identify cell types.
Disadvantages of a manual differential
1. Time consuming (Average time: 2-10 minutes/slide)
2. Standardization (Id and characterization of cells subjective)
3. Prone to error partly 2/2 low cell cound; band counts unreliable
Automated Techniques
Several methods, including impedance, optical, rf, conductivity to count cells, which has great precision except for in plts, esp if dec
- can make 5-part diff, but cannot count bands well, basophils also
- flags can suggest abnormal cell types (blasts, nRBCs, immature grans, which is calibrated to be very sensitive and less spec, thus there are many flags
- Hb measured c cyanohb/ hemiglobulin cyanide (HiCN) method that lyses RBCs, then oxidizes Hb to hemiglobin cyanide (HiCN) which is measured by spectrophotometry (absorbs at 540 nm), detects all Hb except sulfahb (SHb), and lipidemia and paraproteinemia can gively falsely high readings
- Cell counts use impedance counting size; RBCs lysed after a first count to allow better WBC count
- MCV (the mean of RBC size distribution on guassian curve) and RDW (coefficient of variant of the curve; reflects anisocytosis) determined statistically
Flow cytometry
uses the basic Coulter Principle, but with modern improvements to make cell identification more accurate.
FACS- Fluorescence Activated Cell Sorting- In addition to sorting cells by their size and nuclear complexity, fluorescence-tagged antibodies directed against specific antigens on the cell surface (particularly CD markers) are used to more accurately study specific cell populations.
Quality Control
CLIA and JCAHO require at least two levels of control to be run every for 24 hour period on automated instruments
- controls must represent the entire reportable patient range of instrument
- If two distinct sample pathways are used (automated and manual), both pathways must be tested

Aspiration, diluting, sorting, and graphing c automated cell counter



top 4 are scatterplots, bottom 2 are histograms
Hgb-Fe2+ + K3Fe(CN)6 (potassium ferrocyanide) --> Hgb-Fe3+ (methemoglobin)
Hgb-Fe3+ + KCN --> Cyanmethemoglobin
- Read absorption at 540 nm

Reading the diagram:
Any end parameter is equal to the one adjacent divided by the one beyond.
i.e. RBC = HCT = HGB
MCV MCH
Any central quantity is equal to the product of the other two.
i.e. HGB = RBC x MCH = HCT x MCHC
HCT = RBC x MCV
RBC Molecular Structure and Metabolism
Erythrocyte survival and function is dependent on three things:
1) Integrity of the erythrocyte membrane
2) Operational metabolic pathways
3) Normal hemoglobin structure and function
- three essential functions of the RBC membrane:
1) Maintain shape and deformability of the cell
2) Provide support for surface receptors
3) Control permeability and maintain osmotic balance
Biconcave disk shape gives cell high surface-to-volume ratio
- optimal exchange of CO2 and O2 c flexibility and deformability
- RBC membrane selectively controls nutrient and ion passage into & out of the cell, especially glucose, Na+, K+ ions
- Lipids, proteins, ions, and nutrients interact to preserve RBC membrane structure, shape, and deformability.
RBC Membrane composition (by weight):
Lipids – 40%
Carbohydrate – 8%
Protein – 52%
RBC membrane isa lipid bilayer linked to protein cytoskeleton
- Hydrophilic polar heads of the lipid bilayer face the interior and exterior of the plasma membrane and form a barrier between the plasma & cytoplasm; Hydrophobic tails face the intercellular space
- Carbohydrates and proteins contribute membrane structural integrity
Lipid component of RBC membrane is made of non-esterified cholesterol and phospholipids in roughly equal proportions by weight
- Free fatty acids and glycolipids are present in small quantities
Phospholipids - asymmetrically arranged in outer & inner monolayers
- outer monolayer contains Phosphatidylcholine and Sphingomyelin
- inner monolayer contains Phosphatidlylethanol-amine, Phosphatidylserine and Phosphoinositol
FFA (free fatty acids)
Unesterified cholesterol
Functions of cholesterol: Stabilizes the membrane structure, Maintains surface area, Maintains membrane fluidity
Allows passive cation permeability
Glycolipids- Interact with glycoproteins to form RBC surface antigens
An excess amount of cholesterol will reduce the flexibility of the membrane, causing Target cells (Codocytes) and Acanthocytes
- Conditions that cause an increase in membrane cholesterol include:
Lecithin-cholesterol acyl transferase (LCAT) deficiency; Abetalipoproteinemia; Liver disease (Altered ratio of bile salts causes excess membrane, resulting in target cells)
3 types of RBC membrane proteins:
1) Enzymes- involved with metabolic function of RBCs
- discussed below
2) Integral proteins- interior & exterior of cell, piercing lipid bilayer
- May include surface receptor proteins for cell-to-cell communication
- mobile in lipid bilayer, but lateral mobility restricted
- span lipid bilayer and anchor cell membrane to peripheral proteins
Protein Band 3 (AKA Anion Exchange 1)
- 30% of RBC membrane proteins; is involved in ion transport- (energy dependent) requiring large amounts of ATP; Helps regulate RBC volume and homeostasis
Glycophorins A,B,C,E
- 20% of the membrane proteins;
Glycophorin A is major integral protein
Glycophorins carry sialic residue that gives the RBC a net negative charge, positively charged ions in the blood surround the RBCs and prevent them from aggregating, known as the zeta potential
Glycophorins are surface receptors, CD markers
- anything that reduces the zeta potential will cause in increase in RBC aggregation (Heparin affects the zeta potential and thus should not be used for ESR testing!!!!)
3) Peripheral proteins- under inside surface of lipid bilayer face the cytosol side of RBC membrane, forming a mesh-like support for cell membrane
.- form a mesh-like infrastructure beneath the lipid bilayer, but do not penetrate lipid bilayer and do not interact with hydrophobic core
- The mesh-like pattern provides the cells’ cytoskeleton and determines shape & deformability
4 major groups of peripheral proteins:
1) Spectrin is the predominant structural component of the RBC membrane.
- It consists of twisted filaments of alpha and beta dimers
2) Actin is a globular protein that links the filaments together at the ends to form a flexible mesh.
3) Protein Band 4.1 links spectrin & actin to Glycophorin C, an integral protein.
4) Ankyrin (aka protein band 2.1) links spectrin & integral protein band 3
Iinteractions between the peripheral and integral proteins provide a strong, flexible structure that defines the shape of the red cell.
Hereditary defects of RBC membrane proteins
- involve interactions bwt integral and/or peripheral proteins
- either horizontal or vertical:



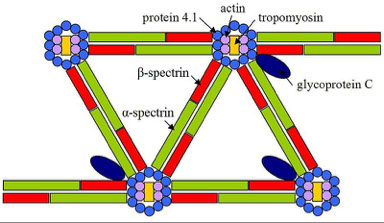
Horizontal interaction (cytoskeleton) defects
Failure of peripheral proteins (Interaction between spectrin dimers, or Interaction of spectrin with Protein 4.1) to work together to form a stable “scaffolding”
- Cells normal until compression in spleen microvasculature
- If destabilization mild, cell may regain normal function and will not be removed by the spleen (as in Common HE)
- If destabilization severe (Hereditary Pyropoikilocytosis), cell becomes fragmented and will be removed by spleen, resulting in anemia
- Result: if cell gets deformed, it can’t regain its shape
3 horizontal membrane protein defects
1) Common Hereditary Elliptocytosis
AD defect in spectrin alpha or beta chain, ankyrin, glycophorin C or spectrin 4.1; up to 1% in Africans
>20% of RBCs are elliptocytes; MCV normal or elevated (due to increased reticulocytes); Reticulocyte count- increased; Osmotic fragility usually normal, mild extravascular hemolysis
2) Hereditary Pyropoikilocytosis
- also thought to be due to a defer in spectrin dimer-dimer interaction
- PB smear shows anisocytosis and poikilocytosis, and abundant RBC fragments and microspherocytes
- this is a severe form of congenital hemolytic anemia
3) Southeast Asian Ovalocytosis

Vertical interaction (membrane) defects
- failure of membrane proteins to stabilize the lipid bilayer
- results in loss of lipid from membrane
- May be result of peripheral protein defects, or defects of interactions between integral and peripheral proteins
Most commonly caused by deficiencies in:
Spectrin, Spectrin and ankyrin, Protein band 3 (AE1), Protein band 4.2
- resulting red cell is osmotically fragile

Hereditary spherocytosis
MC hereditary (AD) hemolytic anemia in Caucasians (1/5k)
- can present as neonatal jaundice or as gallstones / S-megalt in adult
May result in a serious hemolytic anemia
- most cases are heterozygous, homozygous cases are usually fatal
Spherical cells can’t pass through the spleen and are removed
Spherocytes are osmotically fragile
Labs: Reticulocytes inc; MCV variable; MCHC elevated; Osmotic fragility increased, inc retics, extravascular hemolysis + LDH, DAT neg
- Positive autohemolysis test; Varying numbers of spherocytes on PBS
- genes: MC is ANK1 (ankyrin)
- The most sensitive and specific test is the Eosin-5-maleimide binding test (by flow, detects band 3, Rh polypeptides, or CD47, which are reduced in HS)
-- the next most sensitive and specific test is the Acidified Glycerol Lysis test, where RBCs are incubated in a phosphate buffered hypotonic solution with added glycerol, which slows the entrance of water into the cells
--- combining these 2 tests allows for detection of virtually all cases of HS
Hereditary spherocytosis (HS)

Membrane permeability defects
2 forms of hereditary syndromes causing passive permeability of the membrane to cations
- Both usually autosomal dominant, but mechanism of action unknown
- Both involve passive movement of sodium and potassium across cell membrane, yet it is not a defect of the sodium potassium pump.
- In both cases, the passive permeability of cations and water across the cell membrane overwhelms the action of the Na+/K+ pump.
Hereditary Stomatocytosis
AD, Caused by marked influx of monovalent cations (Na and K permeability) followed by water into the cell; causing cells to swell, become stomatocytest
- causes increased osmotic fragility, MCV inc; MCHC dec
- Swollen cells removed by spleen, anemia is benign or mild
Hereditary Stomatocytosis

Hereditary Xerocytosis (HX)
Cause by inc efflux of monovalent cations followed by water out of cell
- Dehydration causes cells to shrink, become echinocytes (xerocytes)
- Cell membrane becomes more rigid, less flexible
- Cells are removed by the spleen
Labs MCV increased; MCHC increased; see Hb condensation at ends of cells on PBS, may resemble HbC or HbSC; Osmotic Fragility dec
- Echinocytes & target cells on PBS
Enzymes
RBC must meet energy requirements to carry functional hemoglobin which can carry oxygen to the tissues for 120 days.
They must also:
Maintain osmotic balance
Maintain membrane integrity
Regulate hemoglobin’s affinity for oxygen
Protect hemoglobin from oxidative damage
- must do all of these things without a nucleus, mitochondria or ribosomes.
- to maintain homeostasis, RBC relies on anaerobic glycolysis.
Energy is provided to all living cells in the form of adensosine triphosphate (ATP), which must be generated by glycolysis
- In RBCs, glycolysis takes place via the Embden Meyerhof Pathway
-- EM Pathway is an anaerobic glycolytic pathway
- provides 90% of the energy used by the mature RBC
- Two molecules of ATP are generated for every molecule of glucose.
ATP is required for cell membrane maintenance
- end product of the Embden Meyerhof pathway is Lactate
--- last step is conversion of Pyruvate to Lactate
- Pyruvate kinase is necessary for converting phosphoenolpuruvate (PEP) to Pyruvate, which generates ATP
A deficiency in Pyruvate Kinase causes less ATP to be made
ATP is required for all metabolic processes
Especially the sodium/potassium pump
The Na+/K+ pump maintains osmotic balance to the cell
Pyruvate Kinase Deficiency
AR; Less ATP made; Cell cannot maintain proper electrolyte gradient
- End result: water is lost from the cells --> cells shrivel, become echinocytes --> Increased membrane rigidity (chronic hemolysis)
- defect in major step in glycolytic pathway (main ATP source for RBC)
Echinocytes are removed by the spleen, patient becomes anemic.
Labs: MCV normal; MCHC normal; Reticulocytes variable; Osmotic fragility normal (May be elevated after several hours (autohemolysis test); Echinocytes may be seen on PBS
- inc 2,3 BPG
- Dx: made by testing for the PK enzyme
In addition to Embden-Meyerhof, 3 other pathways
While the EM pathway generates ATP which helps maintain membrane integrity, other pathways support hemoglobin function, and a defect in these pathways may affect RBC life span
1) Hexose-Monophosphate (Phosphogluconate) Shunt
- maintains glutathione (GSSG) in a reduced state (GSH)
-- GSH destroys reactive oxygen species in RBC, such as peroxide
-- Reactive oxygen species can denature hemoglobin
Denatured Hgb precipitates on inner surface of RBC membrane (Heinz bodies in supravital stained slides), causing membrane rigidity, leads to increased RBC destruction in the spleen
This pathway requires two enzymes:
Glucose-6-Phosphate Dehydrogenase (G6PD)
Glutathione Reductase
- deficiency either enzyme results in oxidative damage to RBCs
Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency
G6PD needed to make NADPH and reduce glutathione for oxidant protection, and RBCs deficient in G6PD sensitive to oxidative stress
One of MC genetic abnormalities; X-Linked recessive; Usually asymptomatic, until patient exposed to oxidative substances (usually by ingestion) - should test for G6PD def when pt not hemolysing (wait 3 mo after hemolytic crisis to avoid false negative results)
- Certain foods are high in oxidative activity: Ingestion of Fava beans causes severe hemolytic episode: this is known as Favism
- also can be 2/2 meds (methylene blue, sulfas, nitrofurantoin) and infx
- Children are much more vulnerable than adults, due to their size
- Favism causes severe hemolysis, c Dark or red urine; Shock
PBS: “Bite” cells (keratocytes) and Heinz bodies (on supravital stain)
- may also see poikilocytes, bite cells, blister cells, and extravascular hemolysis (inc LDH and jaundice)
2) Methemoglobin Reductase Pathway
- main function is to generate NADPH; which is required to reduce Metheme to Heme
- deficiency of NADPH-methemoglobin reductase will cause increased levels of methemoglobin
Hereditary Methemoglobinemia
Autosomal recessive; No RBC morphologic abnormalities
3) Luebering-Rapaport Pathway
- main function is to generate 2,3 DPG
- 2,3 DPG (AKA 2,3 BPG) regulates amt of oxygen released to tissues
- Decreases affinity of Hgb for 02, allowing O2 release to tissues
- no known hereditary disorder associated with the Luebering-Rapaport Pathway.
HX
HX



Heinz bodies in G6PD def
Bite cell in G6PD def


Blister cell in G6PD
Hemoglobin Synthesis & Function
Oxygen is required for aerobic metabolic processes in all living things.
- Oxygen can be carried by plasma alone at rate of 2-3 mL O2/min
-- This is sufficient for lower invertebrates, but the need for oxygen is greater in more complex organisms:
-- At rest, a human requires an oxygen intake of 250 ml/min, and must remove 200 ml/min of CO2.
-- During exercise, that need increases 10 fold.
Hemoglobin in the red cell greatly increases the oxygen carrying capacity of the blood over the oxygen carrying capacity of plasma alone.
Hemoglobin = metalloprotein; functions in delivery and release of O2 to the tissues and to eliminate carbon dioxide.
- responsible for the red color of red blood cells.
- tightly binds oxygen from lungs, carries it from lungs to peripheral tissues of the body where its affinity for oxygen decreases, and the oxygen is released.
The flexibility of the hemoglobin molecule facilitates this change in affinity for oxygen.
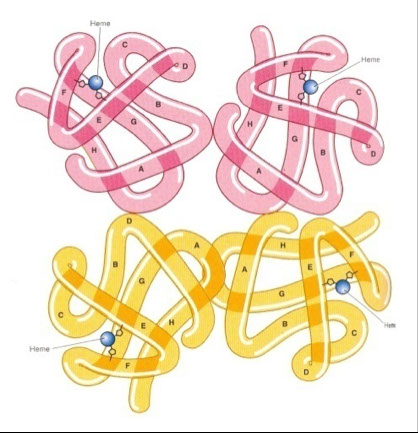
- One molecule of heme is contained within one globin moiety
- Each heme group carries one iron ion, can carry one molecule of O2
- four globin chains per hemoglobin molecule, each carrying one heme. Thus there are four heme molecules in each hemoglobin.
One hemoglobin molecule can carry up to four oxygen molecules
A normal red blood cell carries approximately 280 million hemoglobin molecules!!
Globin is an alpha helix which is structurally rigid.
The tertiary structure of heme refers to the relationship between the globin molecule and the non-protein heme within it.
The quaternary structure of heme refers to how the four globin subunits are linked by non-helical segments.
The non-helical segments in the hemoglobin molecule allow it to be flexible and bendable.
The flexibility of the hemoglobin molecule allows it to have variable affinity for oxygen.
In the lungs affinity is increased so that it can become oxygenated
In the tissues, affinity is decreased so oxygen can be unloaded
The synthesis of hemoglobin takes place from the time the cell is in its earliest maturation stages in the bone marrow through its reticulocyte phase.
There are three important components of the hemoglobin molecule:
Iron
Protoporphyrin
Protoporphyrin becomes heme when iron is inserted
Globin
Normal hemoglobin production depends on an adequate supply and normal function of all of these components
Hemoglobin synth is in RBC precursors through reticulocyte phase
- Like all proteins, globin is synthesized on ribosomes in the cytosol
- Protoporphyrin synthesis takes place in mitochondria and cytosol
- Iron transported to immature cells by transferrin, and inserted into protoporphyrin in the mitochondrion, making it heme.
- body maintains strict control over every aspect of its iron stores, like:
Uptake; Storage; Delivery to RBCs; Re-utilization
- 95% of Iron is recycled
Iron not being used by RBCs is stored as ferritin.
- A small amount of iron is lost by desquamation of ferritin containing cells (1-2 mg/day)
- Large amounts of iron can only be lost through blood loss
The primary use for iron in vertibrates is heme synthesis
- 95% of iron the iron in our body is recycled from old cells and delivered to RBC precursors by transferrin.
- Iron is stored in the body as ferritin, normally in cells of the reticuloendothelial system (RES) of the liver, spleen, and BM
- When either globin or protoporphyrin synthesis are impaired, excess ferritin accumulates in RBCs and their precursors
- Ferritin should not be present in mature RBCs
- ferritin deposits in RBCs on PBS called Pappenheimer Bodies
- if Pappenheimer bodies seen in Wright’s stained slide, the presence of siderocytes should be verified with a Prussian Blue stain
- Ferritin deposits (hemosiderin) are seen in diseases such as sideroblastic anemia, sickle cell anemia, and hemolytic anemia
Siderocytes are non-nucleated RBCs with ferritin deposits that stain positive with Prussian Blue
These are not normally seen in peripheral blood, and may indicate a defect in porphyrin or globin synthesis
Sideroblasts are nucleated RBC precursors with ferritin deposits. These make up 20% to 90% of the normoblasts in the bone marrow.
Ringed Sideroblasts are pathologic ferritin deposits in mitochondria circling the nucleus of normoblasts
- Diagnostic for sideroblastic anemia
- Sideroblastic anemia may be either hereditary or acquired (such as with chloramphenicol use).
Iron Disorders
Iron Deficiency Anemia
MC anemia worldwide, Caused by depletion of body’s iron stores
Reasons:
Increased demand for iron (pregnancy); Nutritional deficiency; Sudden or chronic blood loss;
Hemochromatosis
aka iron overload
- accumulation of iron in body from any cause; Iron gains > Iron losses
-- Total body iron content in normal adults (3-5 g) is the result of the balance between iron losses and iron absorbed from the diet:
- Loss –sweat, shed skin cells, and GI loss at a rate of approximately 1 mg/day (Menstruating adult women lose additional 0.5 to 1.0 mg/day)
- Gain –~10% of the 10 to 20 mg (=1 mg/day) of iron in the diet in affluent Western societies.
- ~2/3 of the total body iron is in the RBCs as hemoglobin, the other 1/3 is stored as hemosiderin
- Hemosiderosis: Iron deposition in organs (liver, pancreas, heart, joints, skin, pituitary gland)
Over years, generation of free radicals generated by Fenton reaction causes tissue damage leading to cell death and tissue fibrosis
dx’d at ~ age 50 men, 10-20 yrs postmenopause women
Tx: Phlebotomy; (Until dec iron, dec % saturation, dec ferritin); Deferoxamine (or other iron chelator) binds iron and excretes via kidney
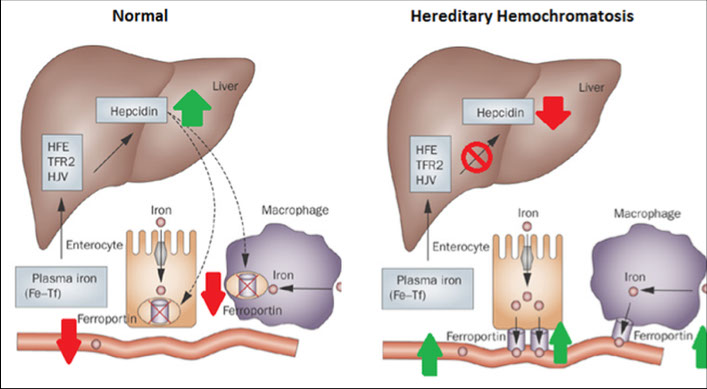
Hereditary Hemochromatosis
aka “primary” hemochromatosis
High Fe (HFE) gene (cr 6) mutation causes altered regulation of Fe by small intestine enterocytes --> Leads to iron overload in blood
- regulates iron absorption
- can be C282Y or H63D mutation
Secondary Hemochromatosis
Not genetic; RBCs circulate for 120 days, after which iron released and recycled by body
Each bag of blood (pRBC) given in a transfusion has 200 mg iron ( = 200 days of normal body iron absorption / excretion!)
African Iron Overload
aka Bantu siderosis or Dietary iron overload; first seen in South and Central Africans
Consumption of large amts of home-brewed beer made in iron pots
Has 46-82 mg/L iron vs 0.5 mg/L normal beer
- Some that don’t drink beer get it too
Ferroportin gene polymorphism (SLC40A1 Q248H)
Hemoglobin Synthesis: Porphyrin
Protoporphyrin synthesis takes place in RBC mitochondria & cytosol
- Protoporphyrin consists of 4 pyrrole rings in tetrapyrrole formation
- precursor to heme is Protoporphyrin IX
Protoporphyrin IX becomes a heme molecule when iron is inserted
The first committed step in the formation of protoporphyrin IX, and the rate limiting step, is the production of δ-Aminolevulinic Acid (delta-ALA)
Delta ALA is the result of condensation of glycine & succinyl CoA in the mitochondria
There are many intermediary products between δ-ALA and Protoporphyrin IX.
An interruption of any of these steps leads to an accumulation of earlier products and cause the porphyrias
Globin synthesis occurs in ribosomes in the nucleated RBC precursor’s cytoplasm
- Residual rRNA in reticulocytes is still capable of making globin
Once a mature erythrocyte has lost its RNA it can no longer synthesize hemoglobin.
There are two matched pairs of globin chains in every hemoglobin molecule, for a total of four chains.
- different types of globin chains determined by amino acid sequence
- The different chains are designated by Greek letters.
- For example, Hemoglobin A (HbA), the most abundant adult hemoglobin, has two α and two β chains, and is denoted as α2β2.
Hemoglobin Globin Chains Frequency
Hb A α2β2 95.0 -97.0 %
Hb A2 α 2, δ2 2.0 – 3.0 %
Hb F α 2, γ2 < 2.0 %
Embryonic, Fetal and Abnormal Fetal hemoglobins
Fetal Hb – Hb F : alpha2, gamma2 (α2γ2)
Embryonic Hb (1st trimester):
Hb Gower 1 : Zeta2, epsilon2 (ζ2ε2)
Hb Gower 2 : Alpha2, epsilon2 (α2ε2)
Hb Portland : Zeta2, delta2 (ζ2δ2)
Abnormal Fetal Hb
Hb Barts : Gamma4 (γ4)
Hb H : Beta4 (β4)
Points to remember:
Embryonic hemoglobins containing zeta and epsilon chains disappear after the third trimester
- The fetal-adult hemoglobin switch occurs around 2 months of age
Detection of HbF - Kleihauer-Betke (acid-elution) test
- HbA elutes from RBCs, but HbF does not, usually see heterocellular pattern in which only a portion of cells are HbF, unless is HPFH
- can also use alkali denaturation technique - HbF resistant to alkali denaturation, HbA gets denatured and precipitated out, then an optical density can show the quantity of HbF
- HPLC can have more accurate HbF quantification
Heinz body test needed to show the unstable nature of hemoglobin H (Hb H), although HPLC and isoelectric focusing and alkaline gel electrophoresis can show a fast-moving hemoglobin
Hemoglobin electrophoresis
Blood lysed on cellulose aceteate at pH 8.6 (alkaline electrophoresis) and subjected to electromotive force
- quantity of Hb variants can be found out by densitometric reading of gel (though not very accurate to measure small quantities of HbA2)
- hemoglobinopathies usually form distinct band of electrophoresis
- acid electrophoresis at pH 6.2 can help id most but not all abnormal Hb
- "Fast" hemoglobins migrate past HbA on alkaline gel (MC are HbH and HbBarts; but can also see fast stuff in hyperbilirubinemia)
- if there's a band in the S region should do a sickle screen (if sickle screen neg must suspect D, G, or Lepore
- thalassemia usually does not produce abnormal bands on electrophoresis (dx'd by thalassemic indices and inc HbA2)
High Pressure Liquid Chromatography (HPLC)
Free of limitation on electrophoresis, can separate HbS from HbD, HbG or Hb- Lepore, and more accurate at quantification
- individual molecules elute at different rates
- light makes deflection on spectrophotometer proportional to Hb variant concentration
- though they cannot separate HbE and HbA2 (done c capillary electrophoresis), HbC and HbO^Arab not easily separated, and bilirubin elutes c Hb Barts
Molecular methods for Hb id
- up to 1/50 HPLC variants cannot be definitively id'd; PCR sequencing can lead to better characterization; esp useful in prenatal dx
The chromosomes where the genes coding for globin chains reside are chromosome 16 and chromosome 11
- Chromosome 16 has two alleles that code for identical alpha chains and one for zeta chains.
- Chromosome 11 holds the alleles for epsilon, gamma (two forms), delta, and beta.
2, 3 DPG controls affinity of each hemoglobin subunit for oxygen
- Hemoglobin exists in two forms; taut (or tense) and relaxed.
-- form it takes depends on presence or absence of 2,3 DPG
High oxygen tension in the lungs makes it more likely for O2 to be added to deoxygenated Hb.
Addition of O2 to one heme group causes that hemoglobin subunit to expel 2, 3 DPG.
- Expulsion of DPG causes salt bridges to be broken, making the space between beta chains narrower
This is the “relaxed” form.
The iron ion moves onto the plane of the heme, which increases the affinity of each additional subunit for oxygen.
This allows cooperative binding of O2 to each additional heme group.
Tissues have a lower oxygen tension than the lungs.
2,3 DPG is produced in response to low oxygen tension.
High 2,3 DPG levels cause it to be inserted between the beta chains, causing salt bridges to form and widening of the space between beta chains.
This causes the heme to move into the taut (or tense) position, causing Hb to have a lower affinity for oxygen
The lower the oxygen tension, the lower the affinity of hemoglobin for oxygen, so more oxygen will be unloaded.
Oxygen saturation curve
2/2 synergy bwt heme molecules, O2 loaded / unloaded very quickly
- As a result, a graph of hemoglobin saturation vs partial pressure (P02) of oxygen is sigmoidal, or S shaped.
- partial pressure of O2 necessary to saturate hemoglobin 50% is called the p50.
- A normal p50 is ~ 27 mmHg
The affinity of hemoglobin for oxygen is affected by the pO2
- If it is low, affinity is high; if it is high, affinity is low.
- The pO2 in the lungs is very high, and lower in the tissues.
A normal p50 is maintained under the following conditions:
- pH 7.4 ; temperature 37.5◦
Metabolic changes which cause these conditions to change will cause the curve to shift to the right or left.
A “shift to the right” occurs when more oxygen is required (hypoxia)
- More 2,3 DPG is made in response to low pO2.
The affinity of hemoglobin for oxygen is decreased.
More oxygen is loaded at a lower pO2.
Factors causing low pO2:
↑ Temperature ; ↑ Acidity ; ↑ CO2 (Haldane effect)
Conditions causing low pO2:
Exercise ; Moving to a higher altitude ; Fever ; Heart/lung conditions
*** CADET face right! *** Inc CO2, Acidity/Altitude, 23DPG, Exercise, Temp cause rt shift ***
A “shift to the left” occurs when there is a high pO2.
Less 2,3 DPG is made.
The affinity for oxygen is increased.
Factors causing high pO2:
Lower temperature ; Alkalosis ; Hyperventilation (decreases CO2); Multiple transfusions with 2,3 DPG depleted blood
Pulse oximetry
2 wavelengths of LEDs [660 nm [red, deoxy Hb] and 940 [infrared, oxyHb]), can estimate arterial oxygen saturation (SaO2)
- cannot measure carboxyhemoglobin, methemoglobin, or sulfhemoglobin and overestimates O2 sat in those settings
Arterial Blood Gas (ABG) analyzers
- calcs % sat after directly measuring pH, pCO2, and PO2
- assumes normal Hb-O2 sat curve, normal 2,3-DPG and no abnormal Hb types
Cooximeter
Multiple wavelenths of light that can specifically measyre oxy- or deoxy-Hb carboxyhemoglobin and methemoglobin
Carboxyhemoglobin
Carbon monoxide
Hemoglobin has a higher affinity for CO than O2
CO will preferentially bind, but will not be released
- Hemoglobin’s affinity for CO is 200 times greater than for O2; and has even greater affinity for fetal Hb
- O2 and CO2 can no longer be carried.
- Reversible at a very slow rate
- Levels of 50% to 80% are fatal
- Smokers levels are 5% to 20%, carboxyhemoglobin absorbs more light at 540 nm, which is why smokers have higher levels of Hb
Sources of carbon monoxide
Household hazards, Boating, Presence of many boats increases CO
- Signs and symptoms of carbon monoxide poisoning:
Cherry red skin and mucous membranes; Headache ; Dizziness ; Nausea ; Vomiting ; Confusion ; Unconsciousness; Respiratory failure
Ultimately fatal
Carbon monoxide poisoning may be reversed if hemoglobin saturation has not reached fatal levels (50%-80%)
Treatment: 100% O2 in a hyperbaric chamber
Laboratory ID c Gas chromatography: Toxic level: 5.0g/dL, must measure c cooximeter, ABG and pulse ox cannot detect
Methemoglobin (Hi, hemiglobin)
result of iron reverting to the ferric (Fe+++) state, instead of usual ferrous (Fe2+) state; is normally 1.5% of total Hb; cyanosis occurs when Hi reaches 10% of total Hb, causing the blood to look chocolaty brown color
- cannot detect Hi c pulse ox or ABG, must use cooximeter
- Hemoglobin with iron in the ferric state has a 3 X increased affinity for oxygen over Hb in the ferrous (Fe++) state
-- Methemoglobin is unable to release oxygen to the tissues.
- Methemoglobin is caused by oxidative stress on RBCs
- The iron in hemoglobin is oxidized, and converted to the ferric state
- Oxidation occurs naturally in the body
-- Methemoglobin reductase (generated by the Methemoglobin reductase pathway) reduces naturally occuring methemoglobin to levels of <1%.
inc methemoglobin may be caused by:
Environmental causes ; Ingestion of strong oxidant drugs ; Exposure to benzene ; Ingestion of nitrites
Hereditary methemoglobinemia
Caused by defect in cytochrome b5 (methemoglobin) reductase or abnormal HbM on which the enzyme cannot act
- pts get cyanosis at 6 mo age
Type 1- “Hemoglobin M disease” - Autosomal dominant ; Mild
Type 2 - Autosomal recessive - Severe, can cause developmental disorders, failure to thrive in infants
Symptoms:
All forms: Bluish skin (cyanosis)
Acquired methemoglobinemia only: SOB, Dizziness ; Headache
Fatigue
Treatment :
Mild cases: Methylene blue- reduces ferric iron to ferrous; Vit C
Severe cases: Hyperbaric treatment; Exchange transfusions
Laboratory ID: Gas chromatography: Toxic level: 1.5 g/dL, or >10% Hb
Sulfhemoglobin (SHb)
Caused by envtl conditions: Sulphur containing drugs ; H2S exposure; also can be 2/2 C perfringens bacteremia (enterogenous cyanosis)
- Sulfhemoglobin cannot carry oxygen: Irreversible condition
-- Sulfhemoglobin containing cells must be removed
Laboratory ID: Gas Chromatography: Toxic level: 0.5 g/dL or 3-4% Hb
- Sulfhemoglobin cannot be converted to cyanmethemoglobin by reference method for measuring hemoglobin
- Heinz bodies can be seen






These are the globin chains found in normal human hemoglobin. The bottom two are embryonic hemoglobin chains and are normally only found in utero during the first trimester.

The chart to the left shows changes in globin chain synthesis during fetal development, birth, and infancy.
- Note that in the third trimester, gamma chain production declines as beta chain production increases.
At around two months after birth, the predominant hemoglobin switches from Hb F (α2γ2) to Hb A (α2β2).



Erythrocyte Production & Destruction
Blood is the body’s most important organ in maintaining homeostasis.
It does this by acting as a medium for transferring heat to the organs, and by acting as a buffer system for the body’s pH.
- Problems with blood composition or circulation can lead to downstream tissue malfunction
- To maintain homeostasis, a healthy erythron must be maintained.
Erythron
The sum of RBCs, acting as an organ
- erythron consists of mature and immature RBCs contained in the:
- Intravascular space- blood vessels
- Extravascular space: Bone marrow ; Organs ; Tissues
Primary function of the erythron is maintain a constant, reliable source of oxygen to the tissues.
- must be maintained by orderly and consistent destruction of old red blood cells which are no longer functional, and the production of new red blood cells to replace them.
A healthy erythron depends on the health of other organs:
Bone marrow - RBCs are produced in the bone marrow
Kidney - Erythropoietin is produced in the kidney
Alimentary system - Essential nutrients: iron, B12, B6, folate
- Absorption of nutrients must be adequate
Spleen - Removes senescent and damaged RBCs, recycles iron
Liver - Site of membrane lipid synthesis ; Secondary site of RBC removal
New RBCs are made in response to erythropoietin stimulation.
- Erythropoietin made in the kidney in response to renal O2 tension
- Hypoxia stimulates EPO release
- Increased EPO =increased RBC production , causing ↑ O2 tension
-- Less erythropoietin is made as more oxygen becomes available
Erythropoietin regulates erythropoiesis in the following ways:
- Causes progenitor cells to undergo fewer mitoses (?)
- Inhibits apoptosis
- Stimulates early release of reticulocytes
- Causes loss of fibronectin, a ligand on endothelial cells which bind reticulocytes in the bone marrow
- Stimulates increase in globin chain synthesis
Total maturation of RBCs is decreased by 3-4 days
Erythropoietin effectors are substances that are known to influence the production or the effect of erythropoietin.
Prostaglandins enhance erythropoietin effect (Prostaglandin E activates production of the CFU-E directly)
Testosterone stimulates erythropoietin production ; Increases responsiveness of immature cells to erythropoietin
Estrogen inhibits production of erythropoietin
Pituitary & Thyroid Hormones can either stim / suppress production and effect of EPO
Erythrokinetics
Disruptions to the balance of the erythron fall into two main categories:
Anemia- RBC destruction exceeds production
Erythrocytosis - production of new red blood cells exceeds the ability of the spleen to remove old cells
The destruction of senescent red cells must be balanced with production of new red cells.
A functional & orderly RBC destruction requires a functional spleen & liver
The normal life span of a RBC is 120 days
As cells age, they undergo physical & metabolic changes:
- Older cells send cytokine signals to macrophages for removal
- Macrophages remove senescent cells in process known as “culling”
- The primary location for removal of RBCs is the spleen
- A secondary site for culling is the kupffer cells in the liver
About 1% of the total volume of RBCs are removed daily.
RBCs must have adequate deformability to pass through spleen
- Aging cells lose this ability
Cells with inclusions are groomed or trapped by the spleen.
- Trapped cells are phagocytized by resident macrophages called “littoral cells”.
- 90% of RBC removal occurs in the spleen. Cells removed include:
Senescent cells
Cells with decreased deformability
Opsonized cells
Cells with inclusions that cannot be removed by grooming
Cells with intracellular parasites (malaria, babesia, etc.)
Cells with crystals (e.g. Hb C) or increased viscosity (sickled cells)
Severely damaged or misshapen RBCs are removed by the liver
The function of littoral cells is to maintain a healthy erythron.
They do this in three ways: grooming, pitting, and culling.
Grooming is the removal from the cell’s surface of excess:
lipids ; cholesterol ; and proteins
Grooming prevents the formation of target cells, stomatocytes, acanthocytes
Pitting is removal of intracellular particles from RBC while leaving the cell intact.
Inclusions removed include:
Howell-Jolly bodies ; Heinz bodies ; Pappenheimer bodies ; intracellular parasites
“Bite cells” may be the result of pitting
Culling is removal of damaged or senescent RBCs, known as erythrophagocytosis
- Aging RBCs undergo morphologic changes that signal senescence.
- Protein Band 3 clusters in cell membrane, appears to be “non-self”
- RBCs are opsonized
Littoral cells phagocytize the opsonized cells.
There are two major factors responsible for age related changes in RBCs that will initiate phagocytosis in the spleen:
Decrease in enzyme activity
ATP becomes less available as EM pathway shuts down
Na + /K + pump less efficient --> More internal Na+; Less internal K +
Greater Internal viscosity
- Phosphogluconate pathway shuts down --> Less G6PD activity causing more reactive O2 species, more unstable hemoglobin
- Luebering-Rapaport Pathway shuts down --> Less 2, 3 DPG made, Oxygen affinity increases
Repeated passes through the spleen
reduces membrane phospholipids - Cholesterol content reduced ; Membrane becomes rigid
- Parts of the cell membrane are removed: MCHC increases ; MCV decreases (younger cells are larger) ; Cells become more spheroidal
- Decreased sialic acid --> Cells become more agglutinable
Spleen also serves as a reservoir for platelets
- 30% of circulating platelets are sequestered in the spleen
-- allows immediate release to the circulation if needed
Splenomegaly (increase in the size of the spleen) can cause increased sequestration of platelets
Causes of splenomegaly: Cirrhosis of the liver; Leukemia or lymphoma
Splenectomy
can occur two ways: Surgery ; Auto-splenectomy
Patients with no spleen have an increased risk of infections, particularly from encapsulated bacteria.
- Peripheral blood findings in splenectomized patients:
Codocytes (Target Cells) ; Howell-Jolly bodies ; Acanthocytes ; Stomatocytes ; Spherocytes
Extramedullary hematopoiesis
When the bone marrow is compromised or overwhelmed and cannot produce enough red blood cells to support the erythron, the spleen will take over erythropoiesis.
Causes: bone marrow infiltration from myelofibrosis or leukemia
- Clinical signs of a compromised bone marrow: Nucleated RBCs ; Teardrop cells
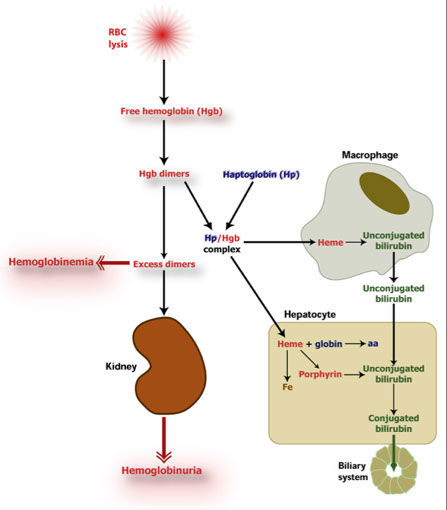
Extravascular and Intravascular hemolysis
removal of RBCs from the erythron, regardless of the cause, is called hemolysis; may occur two ways:
Extravascular hemolysis - Hemolysis in the spleen and liver
Intravascular hemolysis - Hemolysis within the blood vessels
Extravascular hemolysis
The major Normal mode of RBC destruction is extravascular and occurs primarily in the spleen.
About 90% of hemolysis is extravascular.
Macrophages in the spleen ingest old and damaged RBCs
- Hemoglobin is broken down:
Globin is dissassembled to amino acids
Iron is removed from heme which reverts to protoporphyrin
The spleen converts protoporphyrin to unconjugated bilirubin
Iron is picked up by transferrin and transported to the bone marrow
Extravascular hemolytic anemia: G6PD deficiency, Hereditary Spherocytosis, Hereditary Elliptocytosis, Hemoglobinopathy, Thalassemia, Pyruvate kinase deficiency,
- No urine free Hb
- Spherocytes
Intravascular hemolysis
breakdown of RBCs while still in the circulation (see schistocytes).
It is usually minimal and accounts for < 10% of hemolysis
- Degradation products of hemoglobin must be picked up before they can do oxidative damage
Carrier proteins transport them to the liver where they can be further degraded
Intravascular hemolytic anemia: Something wrong with RBC
• MAHA, DIC, TTP, HUS, PNH, PCH, Malignant hypertension, Abnormal heart valve,
- Urine free Hb
- see Schistocytes in PB
The following carrier molecules in plasma quickly capture hemoglobin components for transport to the liver to be recycled:
- Haptoglobin captures Hgb dimers
- Hemopexin captures free heme
Hemoglobin dimers and free heme carried to liver and broken down into bilirubin
Transferrin carries iron to the bone marrow to be used for new RBCs
- If haptoglobin becomes saturated, hemopexin picks up free heme.
- When hemopexin becomes saturated, excess unbound heme appears in the plasma (hemoglobinemia).
- body can efficiently handle intravascular hemolysis of 10% or less.
- If exceeded, may be serious harmful consequences to body
- Haptoglobin becomes saturated when heme dimer concentration reaches 150 mg/dL
- Hemopexin must bind any excess heme once haptoglobin saturated
- Hemopexin becomes saturated at a free heme concentration of 50-100 mg/dL
Free unbound heme may convert to metheme and be bound by albumin, becoming methemalbumin
- Methemalbumin cannot enter the liver, and circulates until more hemopexin can be made by the liver
Consequences of increased intravascular hemolysis: Kidneys
Free heme dimers may circulate through the kidney where they are taken up by renal tubular epithelial cells.
Hemoglobin may be broken down in renal tubular epithelial cells store iron as hemosiderin, but then get sloughed off.
Urine sediment will contain iron (Prussian Blue stain)
The renal tubules can process about 5 gm hemoglobin/day.
Beyond this level, hemoglobin appears in the urine as hemoglobinuria.
Chronic intravascular hemolysis can damage the kidneys
An increase in hemolysis puts a strain on the liver, as it works to convert the excess heme to unconjugated bilirubin
- Albumin bound to metheme is not free to conjugate bilirubin
- Too much unconjugated bilirubin deposits in skin and neural tissue.
- Hemoglobinemia and hemoglobinuria are indicators of excess intravascular hemolysis.
Plasma
Decreased Haptoglobin
Increased Bilirubin (unconjugated)
Hemoglobinemia (free Hb in plasma)
Increased plasma LDH
Urine
Hemosiderin (urine sediment)
Hemoglobinuria (free hemoglobin in urine)
Increased urobilinogen (seen in chronic hemolysis, due to intrahepatic circulation of the breakdown products of bilirubin in gut excreted by kidneys)
Peripheral Blood smear
Schistocytes
(Micro-)Spherocytes

Erythropoietin stimulates the growth of erythroid progenitor cells CFU-E and, to a much lesser extent, of the BFU-E . It contributes to the maturation of all RBC precursors.


RBC morphology
Poikilocytosis
Inc variation in RBC shape; specific to the underlying disorder.
- Poikilocytosis, or “poik”, is rarely reported; it is best to identify the specific type of poikilocytosis present.
- may be due to:
- Inborn membrane or hemoglobin abnormalities
- Mechanical damage to cells passing through the spleen or microthrombi
- Different disease states will cause characteristic abnormalities in the shapes of RBCs
Acanthocytes
cells with irregular thorny projections; Also called “spur cells”
Spherical cells with blunt- or club-tipped spicules
- Irregular sizes
- Placed at irregular intervals
- seen in dz 2/2 alteration of cholesterol content of cell membrane:
Lipid disorders (Hereditary Abetalipoproteinemia); Anarexia nervosa; Malnutrition; IV hyperalimentation, esp. with intralipids; End stage liver disease; Vitamin E deficiency; Post-splenectomy,
- It is also seen in hereditary acanthocytosis
- seen in McCloud syndrome (mutated Kx gene on X cr causes weak Kell antigens)
Echinocytes
Also called “burr cells” or crenated cells
- Reversible, most often occurs in vitro (artifact)
- Do not report artifact- sharper spicules
Shorter than acanthocyte spicules
- More uniform; More evenly spaced around periphery
Causes:
- Artifact - incomplete drying of blood film
-- Interaction of cold blood with glass causes ↑ pH
- Hyperosmolality; as in renal disease
- Actual increase in outer section of lipid bilayer
Seen in:
Uremia; Heparin therapy; Pyruvate Kinase deficiency ; Bleeding peptic ulcers; Hyperlipidemia
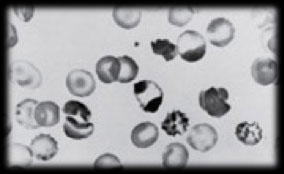
Schistocytes
Cell fragments caused by mechanical damage
Results from cells passing through fibrin deposits
- Alteration of normal fluid circulation can contribute
- Always a signal of a potentially serious condition
- Should always be reported
Seen in:
DIC; MAHA; Prosthetic heart valves; Severe burns; Myelofibrosis; Thrombocytopenic Thrombotic Purpura (TTP); Renal graft rejection
Keratocytes
Also called “blister cells”, “bite cells”, “Helmet cells”
Looks like a bite was taken out of them
Two mechanisms:
1) Inclusion removed by splenic macrophages (bite cells)
- G-6-PD Deficiency ( removal of Heinz bodies);
- Indicates oxidative stress
2) Mechanical damage from fibrin strands
- Microangiopathic Hemolytic Anemia (MAHA)
Dacryocytes
Also called “teardrop” cells
- Tear-shaped or pear-shaped RBCs with rounded ends
- result from mechanical damage to cells
- Infiltration of bone marrow
- Myeloproliferative disorders
- May also result from removal of inclusions
- often seen with microcytic hypochromic cells
- found in moderate numbers in various disease states:
Megaloblastic anemia; Beta-Thalassemia; Renal failure; Heinz body disease; Acquired hemolytic anemia ; Extramedullary erythropoiesis
Target cells
aka codocytes or leptocytes
- Caused by excess cell membrane, and accumulation of hemoglobin in the center and periphery of the cell
- Decreased cellular content (Thalassemia; Hemoglobinopathies; Iron deficiency anemia)
- Increased membrane (LCAT (lecithin-cholesterol acyltransferase) deficiency); Liver disease
Stomatocytes
Caused by electrolyte imbalance, influx of water ; Most often artifact
- Non-artifact stomatocytes seen in:
Hereditary stomatocytosis ; Lead poisoning; Liver disorders ;
Rh null phenotype (very rare)
Macro-ovalocytes
Large, ovoid cells; hemoglobin may be concentrated at the ends
If seen with hypersegmented neutrophils, they are diagnostic of megaloblastic anemia
Cause: abnormal hematopoiesis due to vitamin deficiency
Ovalocytes
Also called elliptocytes, pencil cells, cigar cells
Caused by alterations in cell membrane
Hereditary (Hereditary elliptocytosis; G6PD deficiency)
Metabolic (Iron deficiency anemia; Myelophthisic anemia; Thalassemia; Megaloblastic anemia)
Spherocytes
- Small, uniformly colored; No central pallor; MCHC normal or inc
- Causes:
Mechanical- part of the cell membrane has been removed: decreased membrane-to-vol ratio 2/2 hemolytic anemias or Heinz body removal
- Hereditary : Hereditary spherocytosis
- Artifact : May be seen at periphery of slide; don’t report if artifact!
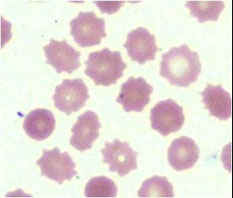
Drepanocytes (Sickle cells)
Elongated cell, sharp & curved at both ends
May resemble elliptocyte, but usually bowed
Cause: polymerization of abnormal hemoglobin when exposed to low oxygen tension
- Often seen with target cells
Diagnostic of sickle cell anemia
Hemoglobin SC disease
- Hereditary hemoglobinopathy ; Double heterozygote, B^s inherited from one parent and B^c from other
- Crystals similar to Hb S, but form only at one end of cell
- More refractile than elliptocytes
- Not as serious as Sickle Cell Disease
- RBCs look “boat shaped”, also taco cells and clam-shell cells and target cells also seen
RBC inclusions
Normal RBCs have no nuclei, and no inclusions in their cytoplasm.
RBC inclusions may be the result of immaturity, or a pathologic process.
Nucleated RBCs
Usually larger than a normal RBC, and contains nucleus
- May have blue-tinged cytoplasm
- Caused by bone marrow stress
Exception- newborns may have nRBCs up to 4 days after birth
- Because of nucleus, hematology instruments count nRBCs as WBCs
- If >5 nRBCs/100 WBCs counted are seen during a differential, the instrument WBC count must be adjusted!!!!!
Formula to correct for nRBCs:
Corrected WBC = (Instrument WBC count) / (100 + # nRBCs counted) X 100
Polychromasia (poly)
Description of RBCs with blue-tinged cytoplasm
- Caused by residual soluble RNA
- Small amount of poly is normal- sign of a competent bone marrow
- Often seen post surgery, or as a healthy response to iron therapy
- Supravital stain may be used to verify that these cells are reticulocytes
Basophilic stippling
Precipitated rRNA which is insoluble
Indicator of abnormal heme synthesis
May be the result of mitochondrial damage
Seen in:
Lead poisoning ; Alcoholism ; Megaloblastic anemia ; Thalassemias
Cabot’s rings are nuclear remnants
Microtubule spindle fibers of nuclear membrane ; Evidence of incomplete extrusion of nucleus ; Extremely rare
May be seen in:
Pernicious anemia ; Lead poisoning ; Leukemias ; Dyserythropoiesis ; Hemolytic anemias
Howell-Jolly Bodies
H-J bodies are nuclear remnants (made of DNA)
- develop during periods of accelerated or abnormal erythropoiesis
- may represent chromosomes separated from mitotic spindle during abnormal mitosis
- Usually only one H-J body is seen per RBC.
Seen in:
Pernicious anemia (B12 deficiency); Post-splenectomy ; Sickle cell anemia (?rare?); Hemolytic anemia
Pappenheimer bodies
Resemble H-J bodies, but there are several per cell
More coarse, irregular ; Tend to cluster at edge of cell
Consist of iron deposits, probably hemosiderin
Seen in:
Splenectomy ; Hemolytic anemias ; Iron overload ; Sickle cell anemia ; Thalassemias
Malaria
Caused by Plasmodium species
Three forms:
Rings ; Trophozoites ; Schizonts
When malaria is suspected, two types of smears are made:
Thin prep - Used to determine morphology
Thick prep - Used to quantify the organisms
Supravital stains
used to stain cells still living
- dye is added to suspension of living cells, the cells actively take up dye
- After incubating the cells with the dye, a smear is made
Differs from Wright’s stain which is applied to fixed cells after the smear is made
Examples of Supravital stains:
Brilliant cresyl blue ; New Methylene blue ; Crystal violet ; Methyl violet
Reticulocytes
When supravital stain is added to living blood cells, it is taken up by the cell
- If a cell has residual rRNA, the dye causes it to precipitate out
- Cells that would show polychromasia on a Wright’s stain have reticulocyte granules with supravital stains
In a manual reticulocyte count,1000 cells are counted
- Cells containing more than two granules are counted as retics and reported as a percentage of the total number of red cells counted
- Because anemic patients have low hematocrits, a smaller number of reticulocytes represent an artificially higher percentage of the total
- Therefore, the reticulocyte count must be corrected in anemic patients:
Corrected reticulocyte count (CRC) = Pt Hct%/ 45 (nml Hct) x Retic %
Absolute retic count = % retics / RBC count
Reticulocyte Production Index (RPI) = CRC x 1/maturation index
- automated technidque are more precise than manual techniques
Heinz Bodies
- cannot be seen on a Wright’s stained smear
Heinz body inclusions are much larger and more round than reticulocyte inclusions
- Heinz bodies formed by oxidative damage to hemoglobin in RBC
Reactive oxygen species (ROS) causes damage to the cells, including hemolysis
- Hemoglobin can’t be reduced, and becomes methemoglobin
- Methemoglobin precipitates, forming Heinz bodies
Causes:
G-6-PD Deficiency ; α-Thalassemia ; Poisoning ; Chronic liver disease
Prussian Blue stain
- Most often done on bone marrow biopsies
- Used to detect abnormal (ferric) iron deposits
- Only ferric iron will stain with Prussian blue
Excessive amounts of ferric iron is found in:
Hemochromatosis ; Hemosiderosis
Ferric iron deposits appear on Wright’s stained slides as Pappenheimer bodies.
Iron deposits stained with Prussian Blue are called siderotic granules, and the cells containing them are called siderocytes
Iron deposits stained with Wright’s Stain are called Pappenhemier bodies
Agglutination is caused by antibodies attached to RBCs, causing them to stick together ; Agglutination is seen in immune reactions
Rouleaux caused by high serum protein content in plasma
- presence of negatively charged protein reduces the zeta potential surrounding RBCs which causes them to repel each other
- As a result, RBCs can get closer together, and form a classic “stack of coins” appearance
Conditions causing rouleaux: Infections; Multiple Myeloma; Cancer; Inflammatory disorders

Poikilocytosis


Acanthocytes

Echinocytes


Schistocyts

Schistocytes
Keratocytes

Dacryocytes

Target cells
Stomatocytes


Macro-ovalocytes
Ovalocytes





Pappenheimer bodies
Howell-Jolly bodies
Cabot ring
Basophilic stippling
Polychromasia
nucleated RBCs
Spherocytes
Drepanocytes (sickle cells)



Reticulocytes
Siderotic granules

Heinz bodies
Rouleaux and agglutination

Hemoglobinopathies
Hemoglobin S
B6 glu --> val
Incidence in blacks is 1/10
- S is a B-chain allele that encodes valine instead of glutamate at position 6 of hb B chain (B6 glu-->val)
Sickle cell trait
- genotype SA
- ~45% HbS, 65% HbA, <3% HbA2
- CBC and PBS normal
- metabisulfite (detects sickling Hb) and dithionate screening tests for HbS are (+), which detect insoluble forms of Hb in a lysate of blood; can also detect SS, SA, SC, SD and C Harlem; false neg if [HbS] is small
- turbidometry test (+)
- generally asx, but inc incidence of hematuria, isosthenuria, papillary necrosis, medullary carcinoma
- exercise induced rhabdomyolysis
- splenic infarcts in hypoxic conditions, like high altitude
Sickle cell disease
- genotype homozygous SS
Short RBC survival, last ~20 days (avg normal is 120 days)
- >80% HbS, 1-20% HbF, 4% HbA2, 0% HbA
-- HbF has inhibitory effect on HbS polymerization (no sx until 6 mo age, when HbS >50%)
- combined sickle cell dz hereditary persistence of fetal Hb (SS-HPFH) is clinically mild
Tx c hydroxyurea raises HbF and dec severity of SS
- hydroxyurea also causes macrocytosis, which can be a useful feature for monitoring patient compliance
Hemoglobins assoc c sickling? *** DOCS *** SD, SO, SC, and SS
Double Heterozygosity
- SC disease has ~1/2 HbS and 1/2 HbC; clinically intermediate bwt SS and SA
- SA / a thalassemia has dec % HbS compared to SA
-- single gene a gene del n (-a/aa) has ~1/3 HbS
-- 2 a gene del (--/aa or -a/-a) has ~1/4 HbS
- SA/B thalassemia has inc % HbS (usually >1/2), dz can be severe
Hemoglobin C
B6 glu--> lys
Hb C trait
- heterozygous AC
- ~1/2 HbC, usually asx, target cells on PBS
HbC dz
- homozygous CC
- 90% HbC, 7% HbF, 3% HbA2, 0% HbA
- mild hemolytic anemia, S-megaly, lots of target cells
- hexagonal or rod-shaped (Washington-monument-shaped) crystals
Hemoglobin E
B26 glu-->lys
Common in SE Asia; clinically asymptomatic
- 2nd MC abnormal Hb worldwide after HbS
- thalassemic indices on CBC and target cells on PBS; elutes at same time as A2 (A2 is elevated)
- usually mild, but coinheritance c B-thal can be severe
- Hb E runs with C on electrophoresis ("A CEO")
Hemoglobins D and G
Pt c HbD and HbG clinically normal
- HbD is B-chain prob, HbG is a-chain defect
- both will run c HbS on alkaline gel
- can see absence of HbS on sickle screen study or on citrate gels
Hemoglobin Lepore
Thalassemic red cell indices
- common in Mediterranean, esp Italy; results from fusion / crossover / knockout of d- and b-genes, causing decreased expression of Hb A2, causing a a microcytic anemia similar to B thalassemia
- runs c HbS on gels, except usually only ~15% of total Hb
- actual HbS rarely present in 15%, so suspect Hb Lepore in that case
- sickle screening tests will also be negative
Hemoglobin Constant Spring (CS)
Thalassemic red cell indices; common in SE Asia; - - / a(CS) a is HbH
- 2/2 mutation in a-globin gene stop codon, making an abnormally long transcript
Hemoglobin Chesapeake and Denver
AD; Hemoglobin variants with inc oxygen affinities, thus a left-shifted oxygen dissociation curve
- pts present c peripheral cyanosis or violaceous complexion
- usually see erythrocytosis in CBC
- only ~1/2 can be detected by electrophoresis
Dx: done by the left-shifted oxygen dissociation curve and DNA analysis


Arrow to hemoglobin C crystals
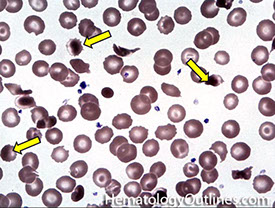


Thalassemia
dec production of normal globin chain, commonly seen in Mediterranean, Africa, and SE Asia
- normally hemoglobin is made up of 4 globin chains, which can be alpha, beta, gamma, and delta
-- HbF = 2 alpha and 2 gamma
-- HbA = 2 alpha and 2 beta
-- HbA2 = 2 alpha and 2 delta
- dec synth of a- or b-chains, causing dec in total Hb prod
- normal amts of unaffected chains causes precipitation, reducing lifespan of cell
-- in a a-thal, b4- and g4 tetramers form
-- in b-thal, a4 tetramers form
- thalassemic indices: microcytosis (dec MCV) and low Hct c normal to inc RBC count; RDW slightly inc to normal (will be markedly inc in Fe def); PBS has microcytosis, target cells, and basophilic stippling
Metzner index = MCV/RBC
- MCV/RBC count ratio <13 favors thalassemia (>15 favors IDA);
Thalassemia minor: Normal serum Fe, TIBC, soluble serum transferrin receptor, RDW, Inc BM storage Fe
from Osmosis B-thal youtube video (https://www.youtube.com/watch?v=oH6SMG3Ykjg)
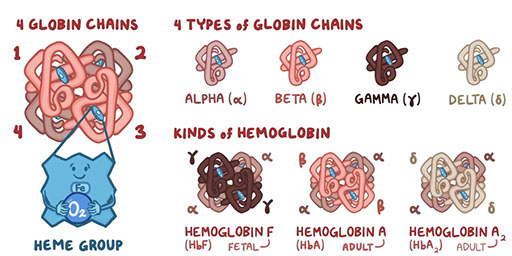
a thalassemia
2 copies of the a gene on each cr 16, for total of 4 a genes
*** 4^2 = 16 ***
- most alleles c thalassemic phenotype 2/2 large structural del
- pathologic haplotypes categorized as a-thal 2 (a+ thal) or a-thal 1 (a0 thal)
-- a-thal 2 (a+ thal) has a normal and a deleted a-gene on cr 16 (-a/), and is the MC genotype in blacks
- a-thal 1 (a0 thal) has 2 deleted a-genes on cr 16 (- - /) and is MC in Asians
- single gene del is asx, c normal CBC and normal electrophoresis
2 gene del cause a-thal trait (alpha thalassemia minor), c thalassemic indices and normal A2%, in absence of Fe def can be interpreted as a-thal trait; ; 5-10% have Hb Bart at birth, can see microcytosis and elevated RBC count
3 gene del causes Hb H dz, c hemolytic anemia and fast-moving HbH on electrophoresis
- assoc c 20-40% Hb Bart at birth, but should only have trace Hb Bart by 1 year of age
- also have normal Hb F. tetramer of gamma chains at birth, hemolytic anemia, and splenomegaly
4 gene del causes Hb Bart dz, c severe hemolysis and Hb Bart on electrophoresis
- manifestations of a-thal are present at birth
"Cis" seen in Asians, 2 alleles knocked out on same cr = worse px
"Trans" in blacks, 2 alleles knocked out on different cr = better px


B-thalassemia
Problems with the B-globin genes in RBCs
- MC in Mediteranean, African and SE Asian populations
AR inheritance, 1 b-gene copy (HBB) on each cr 11, for total of 2 productive genes
- most cases of thalassemia phenotype 2/2 point mutations
- promoter sequence or splice sites can be mutated which can cause either reduced or absent B-chain synthesis
- pathologic alleles can be Bo (no b-chain production) or B+ (dec b-chain production)
- if only one gene of reduced or absent is inherited, leads to B-thal minor
- when two of the reduced genes are inherited, results in B-thal intermedia
- when two of the absent genes are inherited, leads to B-thalassemia major
Sx: does not become evident until 3-6 mo age (due to HbF which has alpha chains and gamma chains) with symptoms manifesting as hepatosplenomegaly, swollen abdomen, jaundice, pallor, anemia, and growth retardation
- B-thal minor is asymptomatic
- heterozygotes (B-thal trait) have inc HbA2 (>3.5%), microcytosis, and normal to inc HbF, though may have normal A2 if pt Fe def
- homozygotes have inc HbF (50-95%), normal to inc HbA2 and little to no HbA
- when there is a B-chain deficiency, free alpha chains accumulate within red blood cells, and form inclusions, which causes hemolysis in the bone marrow, or extravascular hemolysis in the spleen, where it is taken up by macrophages
- hemolysis causes a build up of bilirubin, causing secondary hemochromatosis and jaundice
- hemolysis also leads to hypoxia, causing the bone marrow and extrameduallry tissue to increase RBC production, leading to HSmegaly
- "Chipmunk facies" with enlarged forehead and cheekbones
- skull x-ray can show "hair-on-end" appearance which looks like your arm hairs when you get the goosebumps
If testing for thalassemia in Fe def pts c B-thal, HbA2 may be normal, giving the false dx of a-thal
- RDW is usually increased in both B-thalassemia major and iron deficiency anemia, while it is normal in B-thal trait
- b-thal intermedia and major distinguished by dependence of latter on transfusion
- in db-thal (d and b del) there is normal HbA2 and inc HbF (5-20%)
- in Hb Lepore (fusion of d and b) there in normal quantity of HbA2, slightly inc HbF, and a band in the S region comprising 6-15% (which is the Hb Lepore)
Tx:
- B- thal minor usually does not require treatment
- B-thal major and sometimes B-thal intermedia includes periodic blood transfusions, as well as iron chelating agents such as deferoxamine to avoid iron overload, may also be helpful to get splenectomy
Compount heterozygosity
compound heterozygosity in b-thal and an abnormal b-chain causes inc % of abnormal b-chain; (so in S-b-thal, there is >50% HbS c 1-15% HbF)
- a-thal causes dec % of abnormal b-chains (in S-a-thal there is ~1/3 HbS c 1 a-gene del, and ~1/4 HbS c 2 a-gene dels)
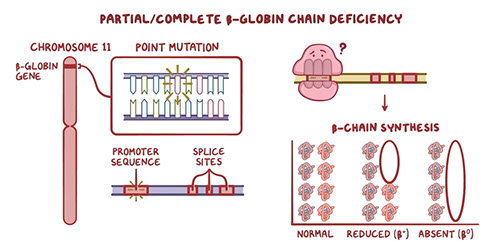
from youtube B-thal osmosis video

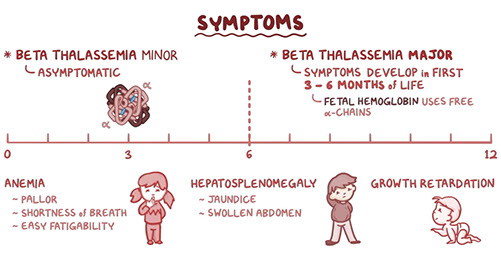



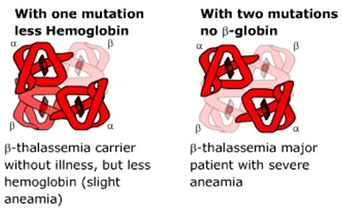
B-thal major

Hereditary Persistence of Fetal Hemoglobin (HPFH)
Normally, up to 40% of total Hb at term is HbF, decreasing to <10% at 6 months, <5% at 1 year, and <1% at 2 years of age
- HbF = 2 sets of dimeric alpha and gamma globins
HPFH is the delayed switch from gamma- to b- or d- chains
- HbF in pancellular districution
- combined sickle cell HPFH seen in ~1% of those c homozygous HbSS, where those affected have ~1/4 HbF and do not get anemia or vasoocclusive episodes
- adult Hb showing HbS, F and A2 can be 2/2 either (1) combined sickle cell HPFH (mild) or (2) combined sickle cell b-thal (severe)
- megaloblastic anemia, aplastic anemia, PNH, Fanconi anemia, JMML and acute erythroid leumkemia can have elevated HbF, but this is usually heterocellular
Warm Autoimmune Hemolytic Anemia (WAIHA)
2/2 warm reacting IgG autoab, which is broadly reactive to Rh ags
- DAT (+) in all cases, c polyspecific and anti-IgG reagents
- see extravascular hemolysis, c spherocytes on PBS
- up to 1/10 hospitalized pts get a (+) DAT, usually w/o hemolysis
- may be primary (idiopathic) or secondary (7/10 cases, usually from cancer [esp CLL], inherited immunodeficiency [CVID, IgA def, Brutons], collagen vascular dz or thymoma)
Cold autoagglutinin disease (CAD) / Cold autoimmune hemolytic anemia (CAIHA) / Cold hemagglutinin disease (CHAD)
2/2 IgM ab c speciicity, MC is anti-I, but can also be anti-i, -H, -Pr, -IH
- can activate complement, so rxns seen at antiglobulin phase c polyspecific antisera
- if monospecific reagents used, cells agglutinated c anti-C3d but not anti-IgG
- if there is hemolysis, it is intravascular
Cold agglutinins can be pathologic or nonpathologic, can predict if pathologic or not based on titer and thermal range (thermal amplitude)
- nonpathologic (b9) cold agglutinins react most strongly at 4C, but can react up to 22C, titers usually <64 at 4C
- pathologic cold agglutinins react over broad thermal range and cause spontaneous agglutination at room temp (more similar to conditions in human body), with titers usually >1000 at 4C
Can be idiopathic (usually in elderly pts, 2/2 monoclonal IgM) or secondary (assoc c infx, like M pneumoniae [anti-I] or EBV [anti-i])
Dx: direct antiglobulin test positive for complement
- may see agglutination on PBS
- clumped RBCs can be counted as a single RBC, falsely lowering the RBC count. Hct is also low.
- Hgb is accurate as RBCs are lysed prior to analysis
- MCHC is high (MCHC = Hb/Hct); thus abnormally high MCHC is a tip off for CHAD
- if the blood is warmed and the tests are repeated, the values will normalize
Tx: targeting of B cells c rituximab or similar meds, or interference with complement coating or erythrocyte destruction
Etiology and treatment of primary cold agglutinin disease. Clonal B cells in the bone marrow produce IgMκ antibodies against the I antigen, which bind to erythrocytes (RBC) at low temperatures. This results in coating of erythrocytes with complement C3b and subsequent destruction by intravascular, but predominantly extravascular, hemolysis in the liver. Treatment strategies include (1) direct targeting of the B-cell clone by rituximab or rituximab-containing immunochemotherapy and possibly B-cell receptor (BCR) or BCL2 inhibitors, or (2) interference with complement coating or erythrocyte destruction.

Paroxysmal Cold Hemoglobinuria (PCH)
Usually in kiddos, secondary to viruses (measles, mumps, chickenpox, mono), though originally described in syphilis
- pts get paroxysmal episodes of hemoglobinuria c exposure to cold
- PBS shows unique intraneutrophilic hemophagocytosis
- tx is to keep that kiddo warm and transfuse prewarmed blood if need be
- 2/2 IgG biphasic hemolysin (Donath-Landsteiner antibody), which has anti-P specificity, and is biphasic bc produces hemolysis only if incubated at 2 different temps in vitro
- DAT positive for C3, with a non-reactive elute, a negative routine indirect antiglobulin test
- Donath-Landsteiner test performed on 2 vials of blood at 2 different temps(4C and 37C), where a (+) test obtained only if incubation of pt RBC at 4C then 37C leads to hemolysis

Donald in a Landrover (Donath-Landsteiner) on the PCH; that is being lanced (lysed) up on both sides (biphasic hemolysin)
PCH - intraneutrophilic hemophagocytosis

Cryoglobulinemia
Cryoglobulins are immunoglobulins that precipitate reversible at low temps, clinically is a systemic immune complex dz c clinical syndrome of palpable purpura (leukocytoclastic vasculitis), arthralgia, HS-megaly, lymphadenopathy, anemia, neuro probs, glomerulonephritis
- blood is drawn and kept at 37C until clotted, then centrifuged at 37C, with serum stored at 4C for 3 days, which is then centrifuged again with anything precipitated being cryoprecipitate, which is sent then for electrophoresis
- in renal bx, MC finding is MPGN type II (dense deposit dz), though can look like thrombotic microangiopathy
- in all tissues, basic pathologic lesion is vasculitis, EM shows subendothelial immune complex deposits c fibrillary or tubular structure in a fingerprint-like pattern
- in PBS, see pale purple cloudy aggs of protein
3 types are recognized: type I and the mixed types II and III
- type I is a monoclonal ig, as seen in MM or Waldenstrom macroglobulinemia
- types II and III affects pts c variety of dzs, such as pymphoprolif disorders, chronic infx, chronic liver dz, and autoimmune dz (SLE) though HCV is the MCC
- type II (MC cryoglobulin type) is mix of monoclonal IgM and polyclonal IgG
-- IgM has rheumatoid factor activity (anti-IgG)
- type III is mix of 2 polyclonal igs
Paroxysmal Nocturnal Hemoglobinuria (PNH)
Acq'd clonal RBC disorder c membrane defects 2/2 dec glycosyl phosphatidyl inositol (GPI) anchors, which normally function to attach array of proteins to the cell surface, which help deflect complement-mediated destruction
- affected cells c low expression of decay accelerating factor (CD55), membrane inhibitor of reactive lysis (CD59) AChE, CD16 and CD48
- GPI encoded by phosphatidyl inositol glycan class A (PIG-A) gene on X-cr; which is sporadically mutated in PNH
Pts classicly described as having episodic nocturnal hemolysis
- MC is to have chronic hemolytic anemia, which cause TBCpenia and leukopenia over time, possibly developing into aplastic anemia or AML
- PNH can also evolve from AML and can present c hemolysis
- may also become thrombophilic and clot
Labs: Hams test (has 6 beakers) and sucrose hemolysis aren't very sens
- in acid serum, hemolysis occurs in both homologous and heterologous serum
- gold standard is flow of PB, which can demonstrate dec GPI anchored proteins on neuts, monos, and RBCs
- for RBC analysis, MC to study CD55 and CD59, which can be classified from type I (normal) to type III (complete deficiency)
- for neuts and monos, CD14/15/16 and 33 usually studied
- if >1/5 type III RBCs or 1/2 neuts, pt at high risk for thrombosis and hemolysis
- Fluorescent aerolysin (FLAER) from Aeromonas hydrophila binds GPI and can be used in place of abs to GPI anchored ags
- lastly, note that LAP score dec in PNH
Quantitative blood cell disorders
Iron deficiency anemia (IDA)
Has overlapping features c thalassemia and anemia of chronic dz (ACD): IDA and thal have target cells and are microcytic, IDA has higher RDW (usually >17), and IDA has pencil cells and prekeratocytes
- MCC in USA is blood loss, worldwide is hookworm infx
- may be 2/2 dec Fe intake or Fe intake inadequate for growth spurts in kids; or in adults from blood loss, dec intake, dec absorption, or inc needs c dec intake (pregs)
PB: dec MCV, dec MCH, anemia, inc RDW, pencil cells, thrombocytosis; BM has dec Fe stores and mild erythroid hyperplasia; serum chem has inc zinc protoporphyrin (ZPP), dec Fe, inc TIBC, dec Fe sat, dec ferritin
- Zinc protoporphorin, which was destined to form heme, accumulates in the red cells in IDA (protoporphorin is also increased in situations where iron utilization is blocked, such as anemia of chronic disease, lead poisoning, and sideroblastic anemia)
Labs: dec ferritin is most sensitive test to dx; most specific test is % iron saturation (transferrin saturation); gold standard is bmbx showing absent Fe on iron stain
- IDA causes HbA2 to be falsely low; in B-thal trait the HbA2 is >3.5%; so concomitant IDA and B-thal trait may cause you to miss BOTH conditions, but especially B-thal trait
IDA

Folate and B12 deficiency
FOlate def = INC homocystein and NOrmal MMA
B12 def = INC homocystein and INC MMA
Folate is cofactor in methyl transfer rxns (converts dUMP to dTMP in DNA synth), and is absorbed in sm bowel from green vegetable intake
- deficiency common in alcoholics, sprue pts, pregs, MTX tx
- labs show inc LDH and indirect bili, dec serum/RBC folate, and inc urinary FIGLU
Folate absorbed in proximal jejunum;
B12 binds R factor in stomach, IF in duodenum, and absorbed in ileum... all organs accounted for in sm bowel
B12 is cofactor for enzymes used in the formation of THF, and def leads to build up of inactive form, methyl folate (the "methyl folate trap"), main source is anumal products
- is bound to R factor in stomach and released from R factor 2/2 action of pancreatic enzymes in the duodenum, where it gets bound to intrinsic factor (IF) where then absorbed in ileum and bound to transcobalamin I and II (TCI and TCII) in enterocytes and released into blood
- labs show in LDH and indirect bili, dec serum B12, inc methylmalonic acid in urine, and dec RBC folate
- can be due to strict vegetarianism, gastrectomy, dilantin tx, TCII def
- HIV infx can cause falsely low B12 levels
- MPNs, liver / renal insufficiency can cause falsely inc B12 levels
PB: ovalactes, hypersegmented neuts, pancytopenia, inc RDW
BM: hypercellularity, megaloblastic changes, erythroid hyperplasia c left shift
Pernicious Anemia
• Autoimmune disease with anti-intrinsic factor and anti-parietal cell antibodies and chronic atrophic gastritis
• Anti-intrinsic factor antibodies (Most specific, but insensitive)
• Anti-parietal cell antibodies (More sensitive, but less specific)
– Positive in other autoimmune diseases
Dx: Schilling test no longer used; antiintinsic factor (anti-IF) ab is sens and spec for pernicious anemia

Anemia of Chronic Disease (ACD)
systemic inflam alters marrow Fe use, causing a normo-normo anemia but can be microcytic as well; usually seen in hospitalized pts
- Fe is stuck in macrophages; hepcidin blocks movement of Fe from macrophages
- assoc c chronic diseases, RA, collagen dz, cancer, chronic infx
- Labs: serum Fe / TIBC normal to dec, % transferrin sat >15%, SSTR normal (vs inc in IDA), Ferritin normal to inc (dec in IDA)
Sideroblastic anemia
Fe in RBC mitochondria from porphyrin abnormalities; in normal individuals, 20-50% of erythroblasts are sideroblasts
- a siderocyte is any red cell with iron, sideroblasts are immature erythroid precursors with iron; both are physiological
See anemia c ringed sideroblasts in BM, PB shows dec MCH c variable MCV (can have bimodal MCV)
- may be acquired (from form of MDS, meds, rads, Cu def, EtOH) or inherited (usually X rec ALAS2 gene defect or in Pearson syndrome)
- Pappenheimer bodies is a basophilic stippling 2/2 Fe accumulation
Labs: inc serum [Fe], inc transferrin % sat, and inc ferritin
Micro: BM c ringed siderblasts and inc Fe stores c erythroid hyperplasia
- abnormal sideroblass include those in which iron granules are increased in number or increased in size or distributed in a ring manner
sideroblasts
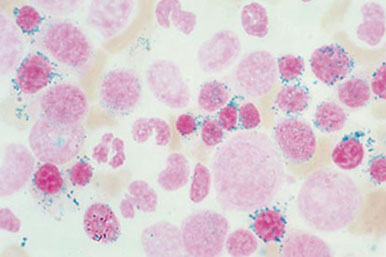
Congenital Dyserythropoietic Anemia (CDA)
CDA II (HEMPAS) is MCC, AR c multinucleated RBC precursors and (+) acidified serum (ham) test (hemolysis c heterologous serum) from failure of normal glycosylation of membrane proteins and is strongly agglutinated by anti-i
- HEMPAS = Hereditary Erythroblastic Multinuclearity with Positive Acidified Serum Lysis test
CDA I is a macrocytic anemia with megaloblastic changes in the BM with lots of internuclear brighing in dysplastic RBC precursors also c inc density of I and i ags, although the ham test is negative
CDA III is autosomal dominant, and shows a mild macrocytic anemia, negative acidified ham test, gigantoblasts in BM and have strong reactivity to anti-i
- CDA III has no assoc c monoclonal gammopathy
Group of rare hereditary disorders characterized by anemia c evidence of ineffective erythropoiesis and distinct BM morphologic abnormalities
Micro: BM shows erythroid hyperplasia, variable internuclear bridging, budding and multinucleation in erythroblasts
• Abnormalities at birth but may not present until middle age
• Splenomegally (types I, II)
• Skeletal abnormalities and dysmorphic features
Labs: Genetic testing for CDAN1 mutations
- SEC23B in type II
– EM for nuclear membrane changes
CDA II (HEMPAS)

Transient Erythroblastopenia of Childhood
Self-limiting normocytic normochromic anemia assoc c hx of prior viral dz, reticulocytopenia, normal RBC adenosine deaminase levels
- plts and WBC usually in normal limits
- median age at dx is 40 mo
Tx: supportive, with RBC transfusion in case of hemodynamic instability
Px: Excellent
Fanconi Anemia (FA)
AR, cr breakage syndrome from mutated FANCA, FANCC, FANCG and inc incidence in white South Africans
- complicated c aplastic anemia, MDS or AML; though macrocytic anemia / TBCpenia present before pancytopenia c inc risk of gastric / cutaneous / liver cancers
- pts usually c absent thumbs / radii, microcephaly, kidney probs, inc HbF, short stature and cafe au lait spots
- screening test involves testing for hypersensitivity of FA cells to DNA crosslinking
Pure Red Cell Aplasia (PRCA)
Acquired in cases of thymoma, collagen vascular dz, meds, lymphoprolif disorders, parvo B19 (gets to erythroid lineage via P ag)
- may be a transient dz in kiddos <4 yo
Congenital form is the rare Diamond-Blackfan syndrome c dec marrow RBC precursors, I ag overexpression on RBCs, inc erythrocytes adenosine deaminase (ADA), inc HbF, and response to roids
Congenital Amegakaryocytic Thrombocytopenia (CAMT)
AR dz 2/2 mutated thrombopoietin receptor (MPL, c-mpl) gene which causes incomplete formation of the thrombopoietin receptor
- usually have inc levels of thrombopoietin
- TBCpenia in neonates, BM has absent MGKCs, progresses to pancytopenia in teens
Tx: BMT is the only cure, and usually need plt transfusions to survive
Congenital Neutropenia (Kostmann syndrome) and cyclic neutropenia
Can be 2/2 variety of inherited causes; MC is mutated ELA2 (neutrophil elastase) gene on 19p13
Sx: neutropenia c recurrent fever, oral ulcers, sinusitis, cervical lymphadenopathy
- cyclic neutropenia / b9 familial neutropenia has intervals of fever c variety of presenting sx, and can cycle every month
- Kostmann syndrome can be seen in neonates c omphalitis that later get pancytopenia up to leukemia
Aplastic Anemia
Lots of causes, most are idiopathic (up to 7/10), while 1/10 from meds, and 1/20 from viruses
- must r/o other causes (MDS, leukemias)
Pancytopenia and hypocellular bone marrow
– Absolute neutrophil count < 500/ml
– Platelets < 20 x 109/l
– Reticulocytes < 1%
– BM cellularity < 25%
• Many causes, but mostly T-cell mediated autoimmune diseases
• Most treatment such as cyclosporin are anti T-cell treatments
• Can develop MDS, PNH or AML
Dyskeratosis Congenita (DKC)
X-linked, rarely AR or AD; is a rare form of ectodermal dysplasia with nail dystrophies in beginning of first decade of life and leukoplakia in the second decade of life
- caused by dec telomerase activity and abnormally short tracts of telomeric DNA compared to normal
- can have aplastic anemia in 1/2 of pts starting in the second decade of life and cancer developing in 1/10 by the 3rd or 4th decade of life
- usually see tbcpenia or anemia followed by pancytopenia, with macrocytosis and elevation of hemoglobin F
WBC Kinetics
- lifespan of WBCs hours/days, tho some lymphs can live for years
Phagocytosis - Monocytes & granulocytes
Antibody production - B lymphocytes
Cytokine production - Monocytes, Granulocytes, T lymphocytes, Natural Killer lymphocytes
WBC disorders
WBC disorders may be either quantitative or qualitative:
- Quantitative differences– an abnormal number of cells
- Qualitative differences – the function of the cells is impaired
WBC disorders may be either malignant or non-malignant.
Malignant disorders are:
Primary- the abnormality is due to a defect in the cell itself
Clonal- defective cells all clones arising from single defective cell
Non-malignant disorders are secondary to another process.
Reactive disorders are secondary to external stimuli
Leukocytosis- Increase in WBCs
Granulocytosis = Neutrophilia, Eosinophilia, Basophilia
Monocytosis
Lymphocytosis
Leukopenia-Decrease in WBCs (by either inc destruction or inc prod)
Agranulocytosis- (complete lack of granulocytes)
Granulocytopenia = Neutropenia
Monocytopenia
Lymphopenia (lymphocytopenia)
Common causes of quantitative changes include:
Primary (pathologic): Neoplasms, malignancies, heme disorders
Secondary: Physiologic (Hard exercise, Stress , Preg, Postsplenectomy) versus Reactive (Acute bacterial / fungal infx, Inflam response, Toxins, drugs)
- cell line responsible for total WBC inc or dec determined by diff count
Kinetics: The production, proliferation, and destruction of cells
Granulocyte Differentiation
- granulocytes = Neutrophils, Eosinophils, Basophils
- CD Markers: CD15, CD45
Differentiation of a stem cell to a granulocyte involves GM-CSF, G-CSF and interleukins, especially IL-3
- (G-CSF) is the principal regulator
Neutrophils
Neutrophils part of Mononuclear Phagocytic System (MPS)
- primary function of MPS cells is to fight bacterial infection in the tissues by phagocytosis of invading organisms
- secondary function is to produce cytokines which facilitate the primary function: Vascular dilation & permeability, Fever (pyrogen), Coagulation (through kinin activation)
Neutrophil kinetics take place in three spaces:
(1) Bone Marrow-Granulopoiesis takes about 10 days
Maturation in bone marrow takes about 10 days
- Two compartments:
Proliferation Compartment (Mitotic Pool or MP)
-- Site of proliferation and maturation of granulocytic precursors
Maturation and Storage Compartment (SP)
-- Non-mitotic granulocytes, Quick mobilization of granulocytes on demand.
Immature granulocytes have CXCR4 ligand (chemokine receptor 4)
Release from bone marrow
- Stromal cells (osteoclasts) have SDF-1 (stromal derived factor), a ligand which binds to CXCR4 that holds neuts in marrow
- Inc G-CSF causes dec stromal cell SDF-1 production, which causes an inc neut release
Circulatory system- In circulation about 7-10 hours
- Intravascular margination, clearance and destruction
Circulating Pool- 50% granulocytes freely circulating in blood
Marginated Pool- 50% granulocytes adhering to endothelial cells
- Half time in peripheral blood is 7-10 hours
- Adhesion to vascular endothelium mediated by integrins and selectins on the vascular endothelium
- Neuts marginate before migrating to extravascular space (tissues)
Neutrophil margination
*RAT SIT!! Rolling Adhesion Transmigration Selectins, InTegrins *
1. Adhesion:
- Bacteria release lipopolysaccharides, and damaged tissue release activating substances
- Release of these substances stimulate tissue macrophages to secrete IL-1 and TNF-α which up-regulate E & P Selectins by vascular endothelial cells
- P-Selectin & E-Selectin on vascular endothelium bind the neutrophil’s L-Selectin loosely (low avidity binding)
- Neutrophil slows down, rolls along the surface of the endothelium in response to bacterial chemoattractants.
- neutrophil sheds its L-selectin and binds to integrin ICAM-1 on vascular endothelial cells (VCAM-1 also involved??)
2. Migration:
- Bound Neutrophils become activated
- Cytokines from neutrophils and mast cells cause vasodilation
-Vasodilation allows activated neutrophils to squeeze between endothelial cells
- Once granulocytes enter the tissue, they cannot re-enter the circulation.
- mediated by integrins ICAM-1 and VCAM-1
3. Opsonization & Recognition:
- Recruited lymphocytes make antibodies which fix complement (opsonins) to invading organisms
- Activated neutrophils in tissues seek opsonin-coated invading stuff
- Phagocytosis is enhanced by opsonization of invading organisms
- Opsonins- mediators which facilitate phagocytosis
- Immunoglobin and complement C3b are opsonins
- Neutrophils have receptors on their surface for opsonins:
-- Fc receptor- recognizes Fc portion of an antibody
-- Complement Receptor- recognizes the C3b complement component
If invading organism opsonized, will be phagocytized more quickly
4. Phagocytosis:
Opsonized bacteria engulfed and digested by the phagocytic cell
1.) Recognition & attachment: Opsonins (IgG and C3) coat target
2.) Engulfment: Pseudopods flow around the particle to be engulfed.
-- Particle is engulfed and fuses with lysosome
3.) Killing/degradation: O2 dependent: Reactive O2 species in lysosomes; O2 independent: Bactericidal permeability agents, lysozyme, MBP, lactoferrin
Tissues- Recruited to site of infection, Phagocytosis, Release of cytokines may lead to inflammation
- Once organism ingested in phagosome, merges with lysosome containing toxic granules.
Neutrophilic granules:
- Oxygen dependent enzymes: Most effective means of killing invading organisms: NADPH Oxidase (Creates the initial respiratory burst which forms oxides & superoxides) and Myeloperoxidase (MPO, Reacts with peroxide and superoxides to produce more toxins)
- Non-Oxygen dependent enzymes: Lactoferrin (Removes free iron), Lysozyme (Reduces pH in the phagosome and oxidizes lps in bacterial cell wall so gelatinase can further degrade it), Gelatinase (Proteolytic enzyme)
Neutrophilia: Absolute value >12.0 x 10^9
An increase in neutrophil populations can be:
Primary - Due to neoplasms (CML), malignancies, hematologic disorders; abnormal immature granulocytic cells seen; LAP dec
Physiologic (Due to: Strenuous exercise, Adrenaline release, No morphologic changes are seen in granulocytes)
Reactive (Acute bacterial or fungal infections, Inflammatory response, Toxins, drugs Morphologic changes are seen in granulocytes)
- usually not >30 bil, can see Dohle bodies, toxic granules, cytoplasmic vacuolation
- reactive change in # granulocytes initially 2/2 mature cells released from marginating pool into circulation.
- Later, BM releases more cells from maturation/storage pool into circulation in response to cytokine stimulation, causing maturation pool to shrink.
- Mitosis in bm increases as demand for more neutrophils increases
- New cells spend less time in the maturation pool and are quickly released to the circulation.
- More immature cells are released from the maturation pool to the circulation, resulting in a “shift to the left”.
Leukemoid Reaction
- reactive quantitative change in # neutrophils above:
>30 X 109 in adults; > 50 X 109 in children
Also see Immature neutrophils (myelocytes, metamyelocytes, bands), and morphologic changes in neutrophils (Toxic Granules; Döhle Bodies; Vacuoles; Degeneration (necrobiosis)
- Causes: Severe infection (most commonly); Toxins (drugs, etc.); Massive trauma; Inflammatory response
Neutropenia: Absolute value <1.0 x 109
- can be different at different lab
Critical Neutropenia is neutrophil value <0.2 x 109
- A critical value is considered life-threatening, and must be called to the physician or attending RN.
- MCCs critical neutropenia (acquired): Increased destruction in peripheral blood (Autoimmune disorder, Drugs [abx, antithyroid, anticonvulsants, procainamide]) or Overwhelming Infection (Viral infections: rubella, rubeola, influenza A & B, RSV), S-megaly
- can also be 2/2 dec prod (large granular lymph leukemia, meds)
Qualitative / morphologic neutrophil disorders
Hypersegmented neutrophils
>6 lobes; Rarely, eosinophils will be hypersegmented
Caused by Megaloblastic anemias:
B12 deficiency; Folate deficiency
There is also a benign hereditary condition which will cause hypersegmentation.
Auer rods
- diagnostic for Acute Myeloid Leukemia (AML); result of fused primary granules in immature cells of the myelomonocytic cell line:
Myeloblasts; Monoblasts; Promyelocytes; Promonocytes
- may be more than one inclusion per cell
Nuclear “twinning” or a nuclear donut
- nuclear lobes look like mirror images, or as one continuous circle.
- induced by chemotherapy or myelodysplastic syndrome
Barr body or Nuclear “Drumstick”
- projection from a nuclear lobe that looks like a drumstick.
It is seen in healthy women and is probably where one unused X
chromosome is stored.
Inherited anomalies of neutrophils
Pelger Huet Anomaly
AD; lamin B receptor mutation; Benign-does not cause disease; Nuclei fail to segment
Homozygotes have round & band forms
Heterozygotes have bands and bilobed nuclei
Lobes are symmetrical
Chromatin is hyper-mature and coarsely clumped
DDx: infx and pseudo-Pelger Huet cells in MDS
Psuedo Pelger-Huet Acquired or “Pseudo-Pelger Huet” when neutrophils mostly bands and bilobed neutrophils; if patient does not have the inherited anomaly can be induced by the following:
Drug ingestion; Leukemia; Myelodysplastic Syndrome; Neutrophil-Specific Granule Disease
- This morphology differs from true Pelger-Huet in the following ways:
Lobes are not symmetrical; Multi-lobed neutrophils may be present; Chromatin is irregular
** Report this as a shift to the left, NOT pseudo Pelger-Huet!
Alder-Reilly Anomaly
AR disorder of Mucopolysaccharide storage and utilization
Also known as: Hunter’s Syndrome or Hurler’s Syndrome
Characterized by dark cytoplasmic granules in the cytoplasm of:
Granulocytes (esp Neuts), Monocytes, and occasionally lymphs
- resembles toxic granulation, but dark granules larger and permanent
- normal neutrophil function
Chediak-Higashi syndrome
Rare, AR, lysosomal defect, has large irregular blue granules in the cytoplasm, the result of abnormal fusion of lysosomal membranes
These granules may be seen in: Granulocytes, NK lymphs, monocytes and are made of primary and secondary granules
- Life-threatening condition!!
Besides abnormal morphology, these patients present with
Partial albinism; Anemia; Neutropenia; Thrombocytopenia; Progressive neural degeneration
- Patients usually die in childhood due to S. aureus infections
- inc risk of EBV+ lymphoproliferative dz and HLH
CHS1 (LYST) gene mutations lead to defective lysosome formation causing dec phagocytosis and defective microtubule polymerization
Tx: BMT is the only known cure
MYH9 related disorders
• Autosomal dominant inherited disorders characterized by large platelets, thrombocytopenia, hearing loss, cataracts and glomerulonephritis
• Formally distinct syndromes with some of the above findings
– May Hegglin Anomaly, Sebastian Syndrome, Fletchner Syndrome, Epstein Syndrome, Alport syndrome
• WBC contains Dohle body and giant platelet; In endoplasmic reticulum/ ribosomes made of RNA
• Neutrophil function is normal
May-Hegglin Anomaly
AD, Cytoplasm of Neutrophils, monocytes, and lymphocytes contain large blue inclusions resembling Döhle bodies, but inclusions are larger than Döhle bodies
- Made of denatured and natural RNA
- Also associated with giant platelets and thrombocytopenia
- Neutropenia may be present
Usually benign, but occasionally there may be mild bleeding or increased infections
Neutrophil anomalies with normal morphology
Chronic Granulomatous Disease (CGD) - X- Rec (MC), also AR
- impaired respiratory burst due to defective NADPH Oxidase
Young men c infx and granulomas at autopsy
- Neutrophils unable to kill bacteria --> recurrent infx; esp. catalase +
Diagnostic Test: Nirtoblue tetrazolium dye (NBT)
- Normal cells test positive-normal respiratory burst reduces the dye
- CGD patient cells test negative- unable to reduce the dye due to lack of respiratory burst
Patients highly susceptible to infection
Early mortality
*** CGD - NBT ***
Myeloperoxidase (MPO) Deficiency
- Autosomal recessive, but complicated; Many mutations implicated
- Most pts probably inherited different mutation from each parent (this is known as compound heterozygosity)
- MPO is deficient in monocytes and neutrophils, causing absent H2O2, and cannot make HCL
- Some patients have decreased ability to fight infections, but most cases are asymptomatic
- Some hematology instruments differentiate neutrophils and monocytes by measuring cellular myeloperoxidase
- here cannot “see” neutrophils/monos, and result in falsely low counts
- Suspect this if machine count is very different than WBC estimate of the smear.
Leukocyte Adhesion Deficiency (LAD)
AR; Very rare- only 300 cases worldwide*; neuts roll but don't stick, no neuts in inflam lesions, though inc neuts in circulation
LAD1: B2 chain of CD11 / CD18 absent ( aka Integrin or Leukocyte Adhesion protein) from the INTGB2 gene on 21q22.23
LAD2: Sialylated oligosaccharide deficient
- Neutrophils are unable to migrate into tissues
- High mortality rate due to recurrent bacterial & fungal infections
- Early bone marrow transplantation is only hope for a cure.
- delayed separation of umbilical cord
Neutrophil-specific Granule Deficiency
AR, Extremely rare, 10 cases in 5 families reported worldwide since 1980
- Neutrophil’s specific granules lack lactoferrin, collagenase, defensins
- Pseudo- Pelger Huet is a distinguishing feature
- Monocytes are deficient or lacking non-specific esterase
Clinical picture:
Increased susceptibility to pyogenic infections
Usually fatal before puberty; Stem cell transplant may cure


RAT SIT!! Rolling Adhesion Transmigration Selectins, InTegrins





Dark toxic granules c high amts of peroxidase; thought to be primary granules
are result of shortened transit time from bone marrow in response to stress.

Döhle Bodies are pale blue inclusions on edge of cytoplasm, made of rough endoplasmic reticulum (ER). sometimes seen with toxic granulation.
Toxic vacuoles look like bubbles in neut cytoplasm,
may be in macrophages,
indicate inc phagocytosis.
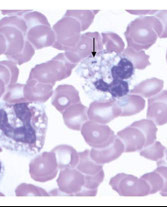
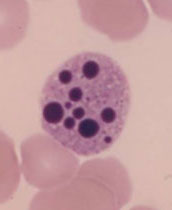
Necrobiosis: In cases of rapid turnover, degenerating neutrophils may be seen.

In cases of overwhelming infection, possible to see intracellular or extracellular bacteria; should be added to the differential report.


hypersegmented neut
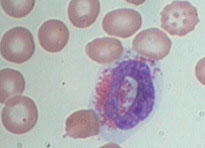
Nuclear donut

-- Auer rod
Barr body-
Alder-Reilly anomaly

Chediak-Higashi syndrome

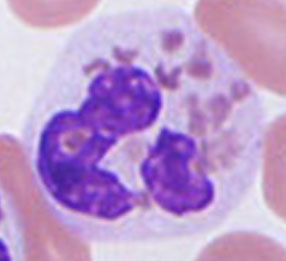

May-Hegglin Anomaly


CGD

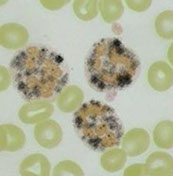
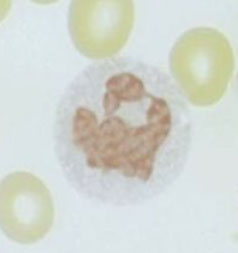
Normal NBT
Abnormal NBT


Eosinophils
Half-life in the circulation: about 18 hours
Mobile and phagocytic
Allergic reactions and parasitic (helminthic) infections
Kill by attaching to surface, degorging granule contents
Granules contain cytokines, proteins, neurotoxin
Eosinophils are the only cells with crystals in their granules (Charcot-Leiden crystals)
Other causes of eosinophilia
Allergic reactions with ↑ IgE- Eosinophils have Fc receptors for IgE
Chronic inflammatory states; Celiac disease; Sarcoidosis; IBD
*** NAACP: Neoplasia, Addisons, Allergy, Collagen vascular dz, Parasites ***
Basophils
Short half life in tissues
Rarely (0-1%) seen in peripheral blood
Similar to Mast cells (but not the same)
Involved in IgE mediated immediate hypersensitivity
Basophilic granules contain histamine, heparin, proteins
Induce production of IgE
Basophils have Fc receptors for IgE
Basophils degranulate after binding IgE, releasing their inflammatory proteins which induce hypersensitivity.
Degranulation of basophils releases chemicals which result in bronchoconstriction, vasodilation, and symptoms of immediate hypersensitivity.
- Basophils also induce lymphocytes to produce more IgE by interacting with CD40.
Basophilia is most often seen in:
Immediate hypersensitivity (Type I):
Allergic rhinitis; Eczema; Hay fever; Asthma; Urticaria;
- It may also be seen in: Viral infx, Chicken pox, Smallpox, Influenza
Monocytes
Monocytes = immature macrophages
- In the peripheral blood 14-100 hours
- May return to the peripheral blood during inflammation
Resident or recruited:
Resident macrophage fixed in tissue
Recruited macrophage - One that migrates to site of active invasion
Monocytes differentiate according to the tissue they become fixed in.
Resident Macrophages:
Kupffer cells (liver); Alveolar macrophage (lung); Microglial (Brain & CNS); Histiocytes (Connective tissue); Osteoclasts (Bone); Langerhans (Skin, tho These are more likely dendritic cells)
Dendritic cells
Dendritic cells may be derived from monocytes or lymphocytes
Dendritic cells are a type of antigen presenting cell (APC)
- Only dendritic cells have the capacity to induce a primary immune response in inactive or resting naïve T lymphocytes.
- During development, develop branched projections called “dendrites”
- Dendritic cells found in all lymphoid and most non-lymphoid tissue
- Before their discovery, immunologists generally thought that macrophages were the only APC in the immune system.
- There are fewer dendritic cells than macrophages in tissues.
Functions of Monocytes / macrophages
Phagocytosis: Phagocytize opsonized particles; Lysosome contents similar to neutrophils.
Monokine production - IL-1; IFN; TNF (cell lysis); G-CSF, M-CSF, GM-CSF (Monokines regulate hematopoiesis and the immune response)
Antigen Presentation - Present processed antigens along with MHC Class II molecules to T helper lymphocytes
- Antigen Presenting Cells (APCs) = macrophages, lymphs or dendritic cells
Clean up: Senescent RBCs; old WBCs; apoptotic detritus; fibrin deposits; Denatured protein; Ag-Ab complexes
Monocytosis (incr in the absolute number of monos) seen in:
Primary (malignant) blood disorders: Monocytic Leukemias (CMML)
Some malignancies may result in monocytopenia
Secondary disorders: Chronic infection (Tuberculosis, brucellosis, listeriosis, subacute bacterial endocarditis, syphilis, rickettsial infections) Recovery phase of acute infections may be a response to neutropenia esp. with tissue damage; Immunologic disorders (autoimmune;SLE, RA, Inflammatory Bowel Disease)
Monocytopenia
Should think of hairy cell leukemia or steroid therapy
- may be omen for impending neutropenia during chemotx
Qualitative Defect of Macrophages
Gaucher Disease
AR; Disorder of lipid storage in monocytic cells; Glucocerebrosidase def results in deposition of lipids in the brain and CNS
- Causes neurologic & other serious complications
Diagnostic marker: Gaucher Cells
Lymphocytes
Interleukin 7 (IL7) is important in differentiation of both B and T Lymphocytes
- 3 basic kinds of lymphs: B, T and NK lymphs
B Lymphocytes mature in bm and express CD19 and CD20
- All B Lymphocytes become either Plasma cells or Memory B lymphs
Plasma cells produce antibodies
Memory B Lymphs carry information for earlier immune response.
Hematogones
• Benign B-cell precursors
• Recovering marrows
– Pediatric
• CD20, CD10, CD34,Tdt+
• Flow cytometry
– Maturation pattern
– Some CD20(+), some CD20(-)
• Versus B ALL: No loss of CD10
T Lymphocytes mature in the Thymus, a primary lymphoid organ
All T lymphocytes express CD3
- two subsets of T lymphocytes, each with a different CD Marker:
CD4: TH - Helper T lymphocytes
Different subclasses of TH cells: Suppressor Ts, or Regulatory Treg
- Secretes different cytokines to activate different cell types
- Participates in Hypersensitivity reactions
- Prevents autoimmunity by suppressing other T cells
CD8:Tc– Cytotoxic T lymphocytes
Cytotoxic T cells are sometimes referred to as “Natural Killer T cells” but are not the same as NK lymphocytes
Kills bugs (mostly viruses) in cytoplasm of cells, and some tumors
Recognizes viral antigens complexed with MHC class I protein (HLA) which is expressed on infected cells
Releases cytotoxins to kill infected cells
Natural Killer lymphocytes maturation site is unclear-probably GALT and thymus
NK lymphocytes do not express CD3, CD19, or CD20
- Because of this, they used to be called “null cells”
CD Markers: CD16, CD56
- % total circulating lymphocytes: 5-15%; Cytotoxic, esp for Tumors
- Can destroy cells without direct antigenic stimulation
- Often appear as “Large Granular Lymphocytes”
- Not the same as NKT cells
Unlike other WBCs, Lymphs never lose ability to proliferate
Because there are different types of lymphocytes, they can vary widely in their appearance, should only be differentiated by surface markers:
Immunophenotyping: use of monoclonal antibodies to classify cells
Lymphocytes important in cell-mediated (innate) and humoral (adaptive) immune responses; they function in the following ways:
Humoral Immune System - Antibody production by B lymphocytes
Cell-Mediated Immune system - Cytokine prod, T lymphs and NK cells
- In addition, Natural Killer lymphocytes have a direct cytotoxic effect on cells in response to interferon.
- Naïve B lymphocytes are undifferentiated and are negative for CD27
- Plasma cells (CD27++) are terminally differentiated- can only produce one immunoglobulin (Plasma cells have a short life)
B memory lymphocytes (CD 27+) can live many years
When B memory lymphocytes encounter the antigen, they quickly differentiate into plasma cells and memory cells.
Plasma cell makes antibody; specific for antigen on organism
- antibody opsonizes the organism, then targeted for phagocytosis
Absolute vs relative changes:
- absolute increase/decrease in lymphocytes indicates the number of lymphocytes has actually increased or decreased.
- relative increase/decrease in lymphocytes is the result of a change in the number of other cells relative to the number of lymphocytes.
(The percentage of lymphocytes relative to the other cells may change, even though the number of lymphocytes is the same)
Lymphopenia
Definition of lymphopenia: Adults < 1000/mm3; kiddos < 2000/mm3
Primary Causes: Aplastic anemia, Leukemia, Hodgkin’s Lymphoma
Secondary Causes: inc utilization (Post acute viral infx); dec synthesis (Immunodeficiency Disease [AIDS, SCID], Autoimmune disorders [SLE]), inc destruction (cytotoxic drugs [Corticosteroids, or rituximab]; Severe stress [Malnutrition])
Lymphocytosis
Definition of Lymphocytosis: Adults: > 4 x 109; kiddos: > 10 x 109
- Primary disorders arise from a defect in a single clone (monoclonal)
- Thus, all defective daughter cells have same appearance, but will be different from normal cells. They are said to be atypical.
- Secondary disorders (reactive lymphocytosis) response to stimulus (virus, transient stress)
- atypical lymphocytes also called Downey cells and activated lymphocytes
- Many lymphocytes from different clones respond, as well as different types of lymphocytes (B lymphs, T lymphs, NK lymphs). These polyclonal cells are said to be variant. (“variant lymphocytes” used exclusively to describe a variation in the appearance of lymphs 2/2 secondary disorder. However, different facilities may use different nomenclature for variant lymphocytes)
- Consistent terminology must be used in order to aid a physician in diagnosing the cause of lymphocytosis.
--Primary causes: leukemia / lymphoma - should consider leukemia / neoplasm in absolute lymphocytosis in adults > 40 yo
--Secondary causes: bacterial / viral (EBV) infx
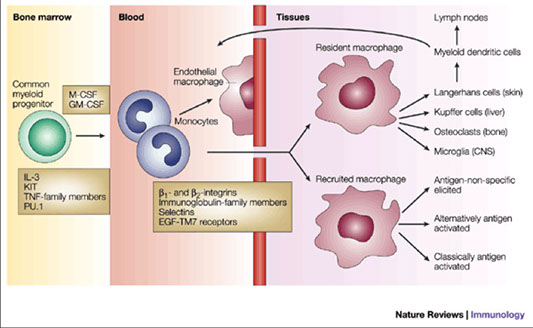


Gaucher dz





Natural Killer cell (not the same as NKT cell)

Reactive lymph

Normal lymph
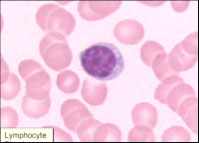
Thrombocytosis
Common finding on CBCs, seen in reactive, neoplastic and spurious causes; usually due to secondary causes
- thrombocytosis with splenomegaly may be assoc c neoplastic dz (MPNs)
Causes of thrombocytosis:
Primary: MPN, MDS with 5q del, MDS/MPN-RS-T and CMML, AML with inv (3), t(3,3), POEMS syndrome, Familial thrombocytosis, Thrombopoietin overproduction, MPL mutations
Secondary: Infx/inflam, Fe def anemia, hemolysis / acute hemorrhage, hyposplenism / post-splenectomy, maliganncy, postop, drugs (TPO, steroids, gemcitabine), "Rebound" following B12/folate def correction
Spurious: Cryoglobulinemia, microspherocytes, schistocytes, bacteria, cytoplasmic fragments of neoplastic cells
Diagnostic algorithm for thrombocytosis
