
Flow Cytometry
History
Basic principles
History
Began in 1980s, with large expensive instruments and poor computational abilities
Improvements made them good at detecting MRD, predict the course of cancers, eval MDS, eval cells for transplant, ID feto-maternal problems, better appreciate cell function, autoimmune dz, fu transplant results in pts c PNH, study microbiology
Lymphocyte Detection
T lymphs have receptor for sheep RBCs, which is CD2 which is the ligand for CD58 (LFA-3)
B lymphs make ig, which used to be detected by fluorescence
- these early studies of lymphs helped learn the normal levels of T and B cells in the PB
- probs c these early tests were that dead T cells did not form good rosettes when aggregating sheep RBCs and binded fluorescent Ig of B cells
- sIg assays were suboptimal bc multiple cell lines have Fc receptors, and autoimmune dz also gave false +'s
- several techniques, like gradient centrifugation and separation of monocytes helped accuracy
- other procedures developed to id monos and exclude them from the cell count
- capping phenomenon where sIg migrate to one side of the unfixed cell and then underwent endocytosis was another problem that obscured B-cell counts
- the Fc and C3 receptors on B cells also helped id
- different receptor types on B and T cells and monocytes developed in labs led to more accurate id
- activated T cell rosettes helped assess cell-mediated immune system (very subjective assay)
- T-mu cells, which had receptor for Fc portion of IgM had single dense granule of reactivity and was found to correspond to T-helper population (not useful today
- B lymphs were detected in tissue sections by dark field microscopy c C3b receptor staining
- before poly-L-lysine, sheep blood would be applied in monolayer in a very subjective technique
Antibody-Antigen (Ab-Ag) Interactions
Dependent on: Ionic forces from charged groups, hydrogen bonding, hydrophobic bonding and Van der Waals forces
- interactions are complex bc have many different antigenic determinants and epitopes
Production of Polyclonal Antibodies
Ag injected into animal with (Freund) adjuvant to immunize animal, and serum specimens taken several months later
- polyclonal reagents preferred, as reacts c more determinants and can react with possibly deficient ags (as in myeloma)
- a prob c polyclonal abs is that they can cross-react and results are inconsistent bc varies bwt batches
Monoclonal Antibody Production
Mouse injected c ag of interest, stimulating B lymphs to make specific abs
- monoclonal abs are more reproducible than polyclonal, and have less cross-reactivity
- monoclonal abs reactivity for 1 epitope can be problematic in precipitation and agglutination reactions
Basic Principles
Flow cytometry is basically the measurement of cellular properties moving in a fluid stream past a set of stationary detectors
- current flow cytometers are the result of a mesh of many scientific disciplines, allowing the analysis of single cells
-- isolated nuclei or debris can be recognized as cells by the flow cytometer, which can be fixed c "gating"
- multiparameter analysis allows eval of multiple features of individual cells
Flow cytometer design
Cells pass single file through laser beam, causing photons to scatter and is then converted to a digital signal
- can be broken down into: the fluidics, the optics, and the computer systems and electronics
Fluidics - cells must be presented to the cytometer in a monodispersed suspension
- a sheath fluid surrounding the sample is forced under pressure through a flow chamber c a conical nozzle that produces laminar flow at a high rate
Optics - most use lasers, rarely arc lamps
- lasers are coherent (all the waves of light are parallel) and monochromatic (of a single wavelength)
- usually use ion gas lasers (argon, krypton); argon is MC bc excites many common fluorochromes at 488 nm; and most lasers are low-power (5-15 mW)
- light from the beam is scattered in all directions, and the light scattered along the axis of the laser is proportional to the size of cell (forward scatter), and light collected from side scatter measures the internal structures (granularity) and membrane undulations of cells (side scatter)
- fluorochromes also absorb and reemit light at lower energies and longer wavelengths, which is emitted in all directions (360 degrees)
- light is passed to different detectors based on filtering and mirrors
Electronics and Computers - detectors are usually photodiodes and/or photomultiplier tubes (PMTs) that produce current when struck by light
- 2 ways to process: peak-sense-and-hold signal processing or integrated signal processing
- these can be amplified in a linear or log domain
- all data collected is stored as list mode data, and is used to form histograms
Cell Sorting
Cells can be sorted while being analyzed by flow cytometry if given electric charge via piezoelecric effect
- some cytometers can take pictures of cells as they are being analyzed
Other techniques which are not flow cytometry are image cytometry (which takes pictures of cells on slides), volumetric capillary cytometry (analyzes fluorescence from cells in a capillary tube), and hematology analyzers (which essentially function as flow cytometers, but don't use fluorescence)
Fluorescence
each fluorochrome has specific exitation and emission spectrum, the difference bwt the 2 is known as Stoke's shift
- not all fluorochromes can be used c all laser, which emit light at a single wavelength
- emission specrums of different fluorochromes should have minimal overlap to not cause false-positives in the analyzers
- MC fluorochrome is fluorescin isothiocyanate (FITC) c emission max at 520 nm
(Electronic / Fluorescence) Compensation
correction of the "bleeding over" or overlab bwt 2 emission spectra
- can be adjusted c modern software
Gating
Electronic window encompassing a given region of distribution delineated by upper and lower limits
- defines a subpopulation in a heterogeneous distribution
- MC gates are CD45 for lymphocyte gait and B-cell markers for light chain expression in different cancers
Forward scatter = Size of cell
Side scatter = granularity of cells

Lymphs (blue)
Monos (purple)
Neuts (green)
Blasts (red)
Hematogones
(aqua)

Compensation
The goal of compensation is to remove the spillover fluorescence of a particular probe from the "wrong" channel (ie fluorescein fluorescence is primarily green, which is measured in the FL1 (FITC) channel but fluorescein also has significant yellow component to the fluorescence that appears in the FL2 (PE) channel)
Mathematically, compensation from FITC to PE simply subtracts a fraction of the FITC signal from the PE signal: the more FL1 (green fluorescein fluorescence) the more spillover into FL2 (yellow fluorescein fluorescence)
ex:
PE_true = PE_measured - 0.15 x FITC_measured
which also works the other way around:
FITC_true = FITC_measured - 0.02 x PE_measured
Exact compensation may not be necessary when determining the frequencies of populations, since there is usually a large valley between positive and negative cells
- compensation is necessary for proper antigen density measurements; any uncorrected spillover contributes artefactually to the measurement
Proper compensation also necessary to distinguish dim populations from negative populations
-- undercompensation results in overestimatung the frequency of dim cells, and overcompensation results in underestimating the frequency
Proper compensation also necessary when there are multiple fluors that spill over into a single channel
Three-color compensation system compensates for spill-over bwt fluorescein and PE (FL1 and FL2) and PE and Cy5PE (FL2 and FL3), which is performed as a 2-color compensation
- thus, 4 coefficients for compensation, 2 for each pair are made
One cannot directly correlate FITC vs Cy5PE, although not too much of a problem bc the ratio of fluorescein is similar bwt FL2 and FL3 to the same ratio for PE
Compensation reference:
1. Compensation. http://www.drmr.com/compensation/
Precursor cells
Hematogones
- Normal B-cell precursors [3]
- Term from 1930s when were first identified in BM aspirates
- less frequently found in PB, lymphoid tissues, but can be difficult to recognize (to differentiate between blasts and lymphs
- easier to identified hematogones by immunophenotype rather than appearance
-- hematogones are B-cells that express antigens assoc c immaturity but include cells at different levels of maturation
- important to recognize and distinguish from blasts, esp residual B-ALL after tx
Steps to identify hematogones:
1. Identify all B-cells, at any stage of maturation
- CD19 is present at all ages of maturation
2. Highlight different stages of maturation, such as mature and immature B-cells
3. Demonstrate orderly, coordinated gain and loss of antigens with maturation
4. Assess for poulations of cells that deviate from the expected maturation pattern
Hematogones [maybe] (circles)

B-cells
- CD19 present throughout all stages of maturation
- sIg expressed on mature B-cells
- CD10 is a more immature marker on B-cells
B-cell maturation
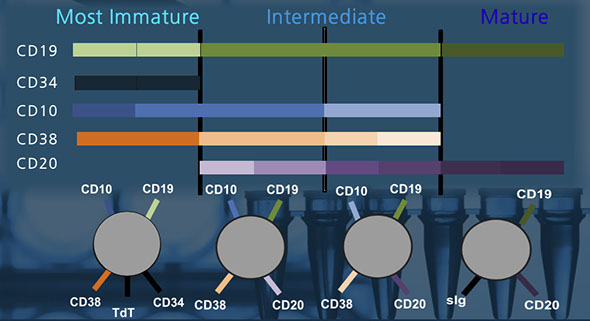
Separating mature and immature B-cells


B-cell markers
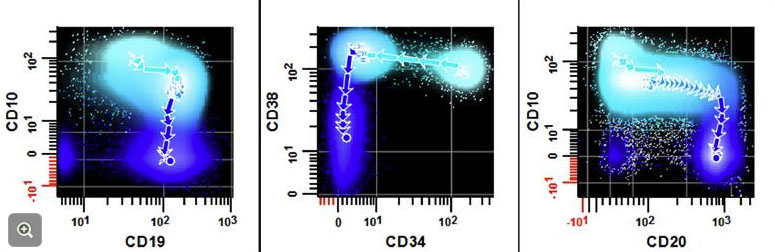
T-cells
Granulocytes
Acute Promyelocytic Leukemia
Does not need to meet 20% criteria!
Flow cytometry is NOT diagnostic - PML-RARa is
- order STAT FISH for PML-RARa, as well as STAT IDH1/IDH2 and FLT3 for new cases of AML and for cases of AML that recur after undergoing remission
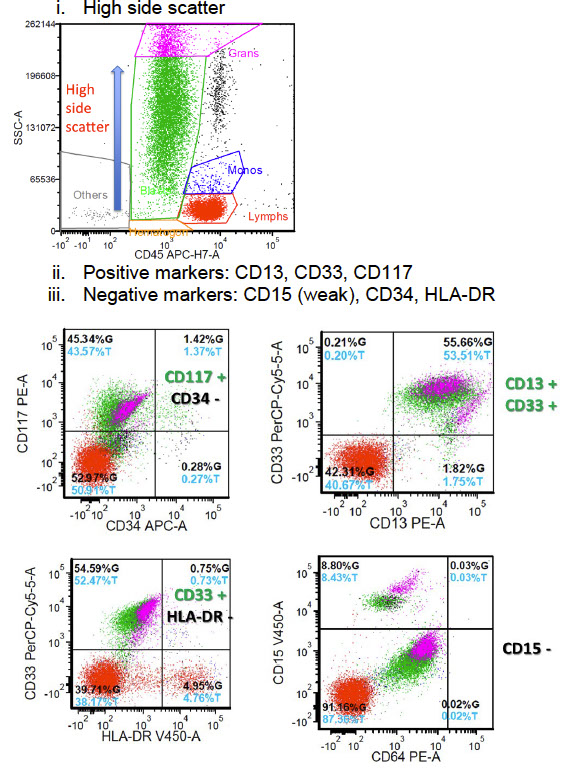
Flow: High side scatter
(+) CD13, CD33, CD117
- negative CD15 (weak), CD34, HLA-DR
APL
References
1. Carey J. Flow Cytometry in Clinical Diagnosis. 4th ed
2. CCEN (Clinical Cytometry Education Network). What is flow cytometry? https://www.cytoed.org/web/e-courses/e-course-What_is_Flow_2/#
3. ICCS E-learning activity: Hematogones. http://www.cytoed.org/web/e-courses/course1/index_html5.html
4.