
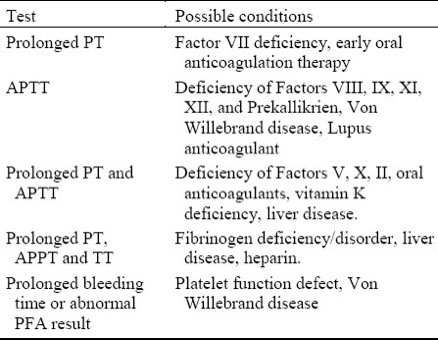
Coagulation
Hemostasis
Primary Hemostasis
Secondary Hemostasis: Coagulation cascade
Fibrinolysis and Anticoagulation
Lab Evaluation of Hemostasis
Hemophilia
Thrombophilia
Therapeutic Agents and Monitoring
COVID-19
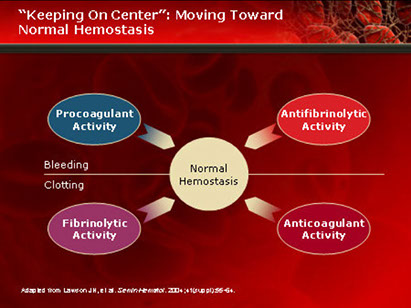
Hemostasis
- balance between procoagulant and anticoagulant
- goal is to maintain a free flowing circulation, but be able to mobilize coagulation as needed to stop blood loss.
3 phases of hemostasis, c 4 major components:
Primary hemostasis = formation of initial platelet plug, and involves the first two components:
- Blood vessels and their supporting components
- Platelets
Secondary hemostasis = stabilization of platelet plug c fibrin and involves the next component:
- Plasma coagulation factors
3rd phase of hemostasis is removal of old thrombi, and prevention of unnecessary activation of coagulation cascade
- sometimes referred to as “teritary” hemostasis
- involves fibrinolytic system.
Primary Hemostasis
Vascular system prevents bleeding by:
- contracting vessels (vasoconstriction) and diverting blood flow from damaged vessels
- Initiating contact activation of platelets and initiating aggregation
- Activating the coagulation cascade.
Intima is innermost layer of vessel.
- made of two layers, the endothelium and the subendothelial matrix.
-- endothelium is layer of endothelial cells in direct contact with the blood flow.
-- most important factor in preventing unnecessary coagulation.
- subendothelium is composed of a matrix created and maintained largely by endothelial and smooth muscle cells.
- endothelial cells of intima is a non-thrombogenic surface which inhibits activation and propagation of coagulation
- enhances fibrinolysis and inhibits platelet agg through the following chemical mediators:
Thrombomodulin, Protein S, Heparan sulfate, TFPI- Tissue Factor Pathway Inhibitor
media and adventitia first response of vessel to injury
- media is made mostly of smooth muscle and elastic lamina, and is responsible for vasoconstriction and vasodilation, and maintaining structural integrity
- adventitia mostly connective tissue, nerves, and nutritive blood vessels called the vasa vasorum.
-- provides connective tissue cell support
-- outermost layer of the blood vessel, and contains highly thrombogenic elements such as collagen.
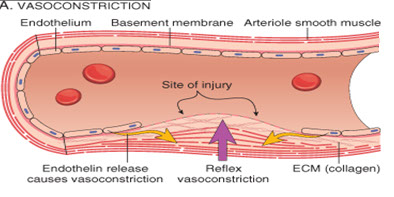
BVs provide body’s first response to injury
- Endothelin is released by damaged endothelial tissue in the vascular smooth muscles of the media layer, which causes vasoconstriction
- Vasoconstriction reduces amount of blood flow through area, and limits amount of blood loss
- Reflexes initiated by local pain receptors in adventitia
-- the greater the damage, the more effective the response
- Vascular spasm is much more effective in smaller blood vessels than in large vessels
Dz's that cause vascular fragility, ie hemorrhagic telangiectasia (Osler-Weber-Rendu) can cause coag probs that resemble plt probs
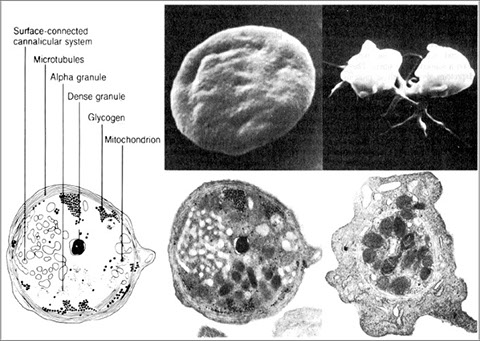
Platelets are cytoplasmic fragments of mkc that bud off mkc in BM and enter circulation
- no nuclei but have structures vital to function
Divided into 4 functional zones:
1) Peripheral zone: assoc c adhesion / aggregation
2) Sol-gel zone: provides plt cytoskeletal system
3) Organelle zone: granules (alpha, dense bodies, lysosomes)
3) Membrane system: dense tubular system where enzymes required for prostaglandin synthesis found
2 major platelet surface receptors:
GP Ib-IX-V is a surface receptor which binds von Willebrand Factor
- Second most abundant platelet surface receptor
- Allows adhesion of platelets at high shear stress (rapid blood flow)
- binding of vWF causes conformational change that allows GPIIb / IIIa to bind to bind fibrinogen
GP IIb / IIIa is the most abundant platelet surface receptor
- mediates platelet-to-platelet aggregation through binding of fibrinogen
Von Willebrand Factor is large multimeric protein present on the surface of endothelial cells
- It is also produced by megakaryocytes and present in alpha granules of platelets
Within the platelet are membrane compartments:
The Open Canalicular System (OCS), or Surface-Connected Cannicular System acts as a conduit system for entrance of plasma substances to and release of granule contents from the platelet.
It is also provides additional membrane capacity during activation
The Dense tubular system (DTS) consists of microtubules, and is a major site of calcium storage.
- plays role in thromboxane and prostaglandins synth
- analogous to smooth endoplasmic reticulum in nucleated cells.
Platelets contain 2 types of granules:
1) Alpha granules contain proteins involved with platelet adhesion, cell-cell interactions, and vascular repair
2) Dense granules contain secretable factors which primarily activate or recruit other platelets (aggregation), known as the platelet agonists
Alpha granules
- Soluble adhesive proteins: fibrinogen; fibronectin; thrombospondin; B-thromboglobulin; von Willebrand Factor (vWF)
- Enzymes & inhibitors: Plasminogen; alpha2-Antiplasmin
- Procoagulants: Factor V; Platelet Factor 4(PF4)
Membrane-bound adhesion proteins: P-selectin
*** Knowing what is in alpha granules has little VAlue For AP *** alpha granules have Alpha2-antiplasmin, P compounds (PF4, P-selectin, PDGF), Fibrinogen / Fibronectin vWF / factor V ***
Dense granules
ADP (cause vasoconstriction); Serotonin; CA++; Polyphosphate; Arachidonic Acid
- Membrane-bound adhesion proteins: P-selectin, Granulophysin
*** the dense granules are dense b/c it is packed with small stuff (small molecules)***
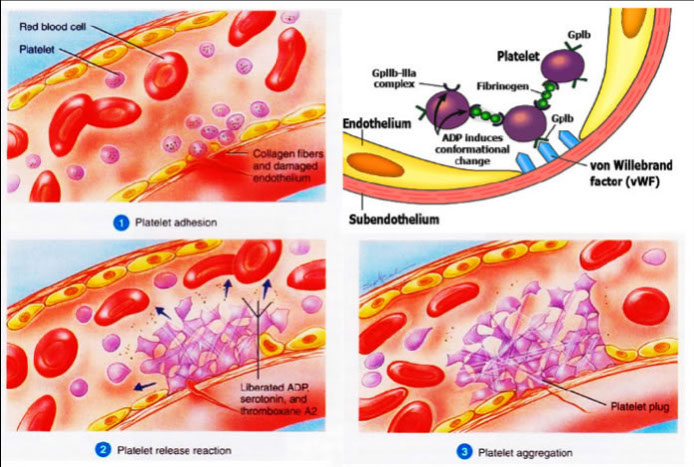
Platelets initiate hemostasis in 4 steps:
Step 1: Adhesion
Platelet adhesion begins exposure of subendothelium
- Subendothelial matrix proteins (such as collagen) are ligands for platelet receptors
- Exposure of proteins --> rapid adhesion of platelets
- von Willebrand Factor secreted by plts+endothelium
- vWF forms additional links between collagen and platelet receptor GP Ib
- GPIa-IIa is receptor for collagen, and Bra/Brb ag
- additional links allow adhesion at high shear rates
- GPIb activated by ristocetin in vitro, shear forces in vivo
- GPIc/IIa is a fibronectin receptor
- GPIb/V/IX (CD42) complex on plt cell surface, which is a receptor for vWF
Step 2: Aggregation
Aggregation begins when platelets undergo a shape change from discoid to spiny sphere, exposing fibrinogen receptors
- GP iib/IIIa receptor binds fibrinogen
- Spiny protrusions increase the surface contact area with the endothelial surface
- induced by early release of Ca2+ and ADP from dense granules
- Aggregation prior to the release phase is reversible
- GP iib/iiia inhibitors (Abciximab-Reopro, Integrilin, Aggrastat)
Step 3: Activation and Release
- shape change compresses the platelet, leads to further degranulation, become activated c release :
1) dense granules - calcium, serotinin, and ADP, ATP
2) Alpha granules release fibrinogen, fibronectin, PDGF, P-selectin, platelet factor 4 (PF4), and vWF
3) Arachidonic acid is released, which is converted to thromboxane (Thromboxane A2- promotes vasoconstriction & aggregation)
Dense Granules
Serotonin- promotes vasoconstriction and acts as a secondary agonist
ADP- acts as a platelet agonist and promotes platelet activity
- Release of these substances expose surface receptors and results in irreversible aggregation.
Degranulation stim by plt agonists, like epi, ADP, thrombin, plt activating factor (PAF), collagen, TXA2
Platelet plug formation
Release of granule contents increase the affinity between Platelet receptor IIb/IIIa and fibrinogen
Platelets become joined via a fibrinogen bridge
Fibrinogen forms cross-links with Platelet receptor IIb-IIIa
End result: platelet plug
A platelet plug can become a stable clot when fibrinogen is converted to fibrin via the coagulation cascade
Step 4: Clot stabilization
Once degranulation has finished, the platelet becomes a “spreading disk”
- Aggregation irreversible if plts in this conformation
- platelets release factor V and expose platelet factor 3, accelerating the coagulation cascade
- eventually stabilizes platelet plug into a stable clot
Platelet activation when no injury is present leading to thrombus formation is the leading cause of death in the western world
- Myocardial Infarction, stroke and pulmonary embolus are all almost invariably related to venous and/or arterial thrombosis
- Aspirin dec likelihood of thrombus formation
- does this by interfering c Arachidonic pathway
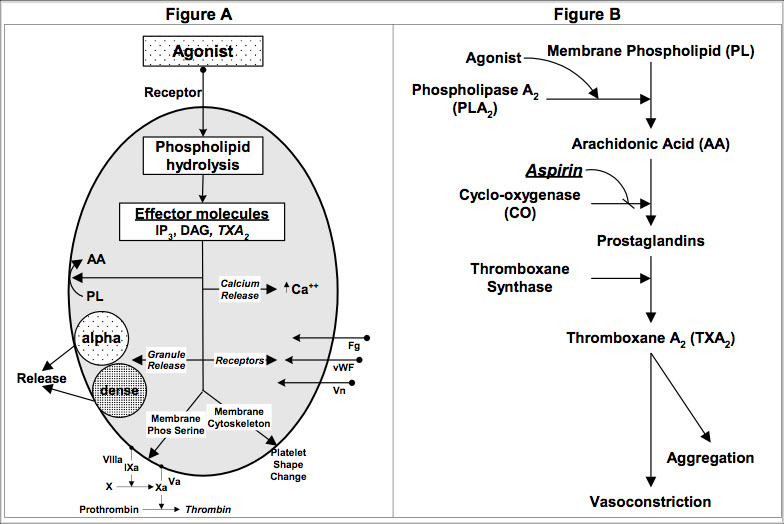
The Arachidonic Pathway
During the activation phase, platelets are stimulated by agonists which cause hydrolysis of membrane bound phospholipids converting them to arachidonic acid.
Arachidonic acid is converted to prostaglandins by cyclo-oxygenase.
Prostaglandins are converted to thromboxane (TxA2), a potent platelet aggregator and vasoconstrictor.
Aspirin irreversibly binds to cyclo-oxygenase, making it non-functional for the life of the platelet.
This decreases activity of TxA2 by 95%.
Von Willebrand Factor (vWF) is important in coagulation in general and platelet function in particular.
It is a large, multimetric protein secreted into the extracellular matrix from endothelial cells
It is also synthesized in megakaryocytes and stored in platelet alpha granules.
VWF facilitates platelet adhesion to the endothelium by binding to platelet surface glycoprotein Ib/IX, especially at high shear rates.
vWF serves two functions in coagulation:
Helps platelets adhere to thrombogenic surfaces under high shear rates
Acts as a carrier for coagulation factor VIII, which is an essential cofactor in activating factor X.
Hereditary conditions involving vWF are the most common bleeding disorders.
Structure of von Willebrand Factor
- basic subunit of vWF is a dimer which is synthesized in the endoplasmic reticulum of endothelial cells and megakaryocytes.
Dimers are sent to the golgi where they polymerize.
- finished molecule consists of repeating structures of variable length, with MW up to 20 million
- Each subunit of vWF has many binding sites, including:
GP1b-IX, important for platelet-collagen binding
GP IIb-IIIa- Fibrinogen receptor
Two sites for collagen binding
One site for heparin binding
Binding site for factor VIII
The structure of the blood vessel is responsible for its anticoagulant and procoagulant properties.

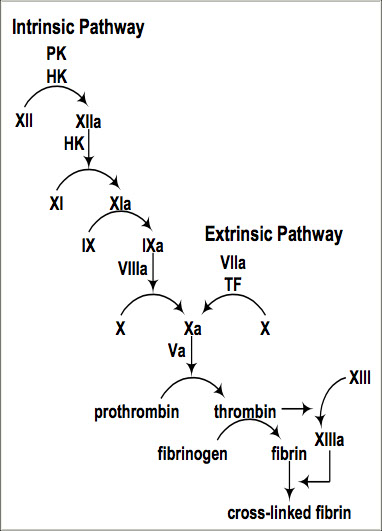
Secondary Hemostasis:
the Coagulation Cascade
A complicated way to form fibrin
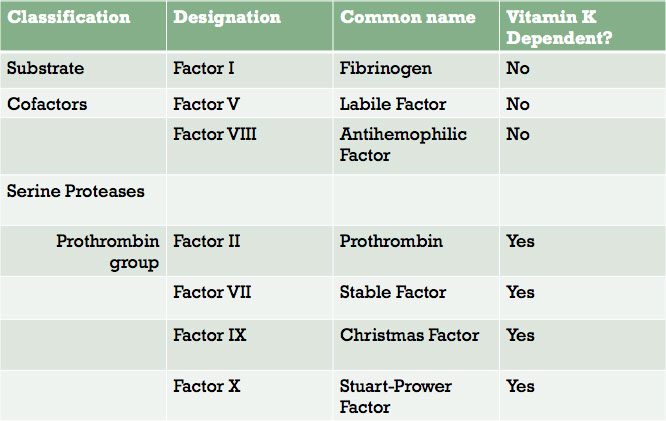
- coagulation factors are soluble proteins that circulate in plasma in inactive state.
- Most coagulation factors designated by Roman numerals, but they also have common names.
- The numerals were assigned in the order in which they were discovered, not the sequence in which they appear in the cascade.
The coagulation factors are mostly classified as serine proteases and coenzymes
- a protease is an enzyme that cleaves an amino acid chain one or more times
- A serine protease has the amino acid serine at its active site
A coenzyme is a cofactor of an enzyme reaction
All of the coagulation factors and vitamin K are synthesized in the liver. Factor VIII is also synthesized in endothelial tissue throughout the body.
Terminology
Zymogen: An inactive precursor of an enzyme
Activated factor: A zymogen with part of its chain cleaved off, so that it becomes a proteolytic enzyme
- When a coagulation factor is in its zymogen form, it is designated by its Roman numeral only
-- Example, Factor X is the zymogen, or inactive, form
- When coagulation factor is activated, designated with the roman numeral and small “a”, for “activated”.
-- Example: Factor Xa is an activated enzyme
The next two slides list the coagulation factors in table form, grouped according to their function:
Pathways of Coagulation
The classical model of coagulation divides the coagulation cascade into two separate pathways converging into a common pathway:
Extrinsic Pathway, which consists of Factor VIIa and Tissue Factor; is so named because TF is “extrinsic” to the vascular system
- Extrinsic Pathway is initiated when clotting factors in the blood are exposed to Tissue Factor (aka factor III)
Tissue Factor activates Factor VII, and becomes the TF/VIIa complex.
The TF/VIIa complex converts small amounts of Factor X of the common pathway to Xa, which is the beginning of the common pathway.
- VII is unique to extrinsic pway (and is the "tense" of the extrinsic pway)
Intrinsic Pathway is so named because all factors arise within the intravascular domain
- test factors XII, XI, IX, VIII, prekallikrein, HMWK
- tenase complex: made of Ca2+, VIIIa, IXa on plt surface, catalyzes fX to fXa
The Intrinsic Pathway begins with the conversion of Factor XII to XIIa by negatively charged surfaces, such as glass.*
XIIa activates XI to XIa
XIa activates IX to IXa
IXa, along with cofactor VIIIa, converts X to Xa, beginning the common pathway
* Important note: Factor XII is not necessary for in vivo coagulation, and a deficiency of Factor XII will not result in a bleeding disorder.
Common Pathway
- Factor Xa with its cofactor, Va, convert prothrombin (Factor II) to thrombin (Factor IIa)
- Thrombin is the enzyme which converts fibrinogen (Factor I) to fibrin in the prothrombinase complex (akin to the "tenase" complex of intrinsic pway) by cleaving the alpha and beta chains, which releases the N-terminals (fibrinopeptides A + B) and exposes their "sticky ends"
- spontaneously forms fibrin molecules that form protofibrils that form side-to-side and are ultimately stabilized by f XIII
Factor XIII, activated by thrombin, further stabilizes fibrin by causing cross-links between fibrin molecules.
- plasmin degreades fibrin
- tissue Plasminogen Activator (tPA, release from vascular endothelium) converts plasminogen to plasmin
The classical coagulation cascade model was formulated by in vitro analysis of coagulation in the laboratory
- In vitro laboratory analysis of coagulation occurs in a test tube using platelet poor plasma.
-- Advantage: the intrinsic and extrinsic pathways may be evaluated separately.
-- Disadvantage: The classical model does not accurately reflect what happens in vivo
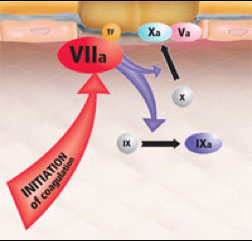
Cell Based Model of Hemostasis
In vivo coagulation takes place in the presence of exposed tissue factor bearing cells and platelets, both of which are absent in in vitro testing, and which contribute significantly to the formation of a stable clot.
For this reason, a cell-based model of hemostasis was proposed to better explain the interaction of platelets and tissue factor bearing cells with the coagulation cascade in vivo.
In the cell-based model, hemostasis occurs in three overlapping phases:
Initiation phase
- initiation phase begins on a tissue factor bearing cell
TF is normally expressed in the adventitia of blood vessels, and by epidermal, stromal and glial cells.
When these cells expressing TF are exposed to blood at the site of injury, the TF binds Factor VII.
Once bound, Factor VII is rapidly converted to VIIa
The VIIa/TF complex, localized to the site of injury, catalyzes two reactions:
Activation of factor X to Xa
Activation of IX to IXa
Role of Xa formed on TF-bearing cells
Factor Xa interacts with its cofactor Va to form the prothrombinase complex, and generates a very small amount of thrombin (IIa) in the vicinity of TF-bearing cells.
The localization of factor activation to the site of injury helps to limit hemostasis
By itself, the thrombin generated by this process is insufficient to convert fibrinogen to fibrin, but it acts as a primer to the coagulation system
The thrombin formed in the initiation phase primes the coagulation system in the following ways:
Activates platelets to release more agonists, recruiting more platelets to the site of injury.
Releases factor VIII from von Willebrand Factor and activates it.
Plays a role in activating Factor V
Activates Factor XI, initiating the Intrinsic Pathway
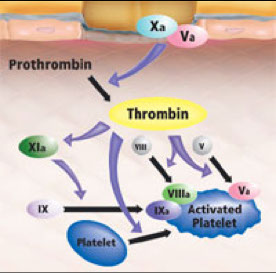
Role of IXa- Amplification Phase
Factor IX is activated to IXa on activated platelets in close proximity to the TF-bearing cells.
Unlike Xa, IXa can diffuse to adjacent surfaces because it is not inhibited by TFPI and is only slowly inhibited by AT, as compared to Xa.
On the surface of the activated platelet:
IXa forms a complex with its cofactor, VIIIa, which is known as the “tenase” complex.
Specific sites on activated platelets bind IXa and promote formation of the tenase complex.
Once “tenase” is assembled, factor X is recruited from the plasma and activated to Xa on the platelet surface.
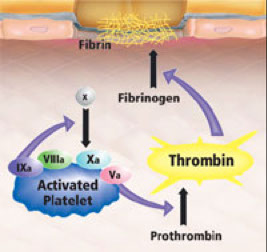
Propagation phase
Xa then associates with Va on the surface, forming the so called “prothrombinase” complex.
This “prothrombinase” complex acts to generate large amounts of thrombin (IIa) at the site of the platelet plug where fibrinogen is already present, converting it to fibrin.
Factor XIIIa, activated by thrombin, cross-links the fibrin and stabilizes the plug.
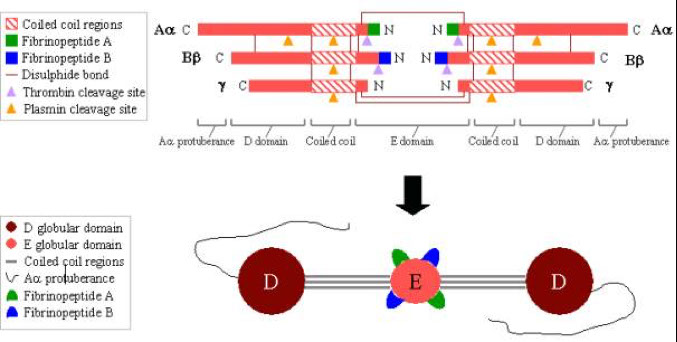
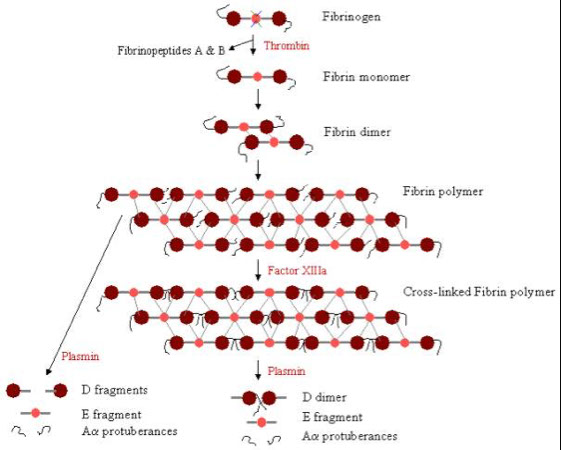
Fibrinogen
- links platelets early in platelet plug formation
- Fibrinogen is converted to fibrin and forms the structural network that consolidates the platelet plug into a solid hemostatic clot
- fibrinogen is a trinodular molecule containing two outer D domains, and one central E domain.
-- To convert fibrinogen to fibrin, thrombin cleaves fibrinopeptides A and B (blue and green) from the E domain
Polymerization of fibrin
With the release of fibrinopeptides A and B, the fibrin monomers align themselves in a staggered half overlapping orientation, held together by D-E contacts.
The 2-stranded protofibrils polymerize by forming lateral, longitudinal, and transverse cross-links between D-E contacts.
Factor XIIIa, in the presence of calcium and thrombin, causes cross-linking of the fibrin polymers between the D domains, which stabilizes the fibrin polymer.
Fibrin is degraded by plasmin between the D and E domains
-Plasmin degradation of cross-linked fibrin results in E fragments and D dimers (both are Fibrin Degradation Products or FDP).
Plasmin degradation of fibrinogen results in D fragments and E fragments.
Therefore, the presence of D-dimers along with FDPs indicates the breakdown of fibrin, while the absence of D-dimers in the presence of FDPs indicates the breakdown of fibrinogen.
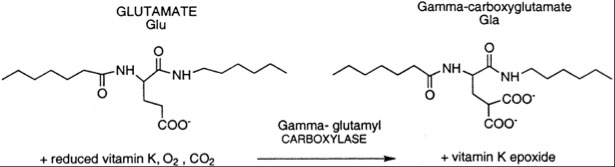
The Vitamin K dependent coag factors contain many gamma-carboxyglutamyl residues (Gla)
During the synthesis of these proteins, Glutamic acid is converted to -carboxyglutamyl residues by gamma-glutamyl carboxylase.
This reaction requires CO2 and reduced vitamin K.
Carboxylation of Glutamic Acid to Gla allows calcium-ion binding within the Gla domain.
Binding of calcium to Gla residues causes a conformation change in the protein that exposes hydrophobic amino acid residues which then mediates association to phospholipid membranes
In other words, calcium bound to the Gla domain of a vitamin K dependent coagulation factor mediates its attachment to the cell membranes, especially platelets
This is important to the coagulation reaction because:
It localizes the reaction to the cell surface (especially the platelet surface), helping to control the clotting process
Phospholipid binding also increases the proteolytic activity of Gla-containing proteases
For each glutamyl residue that is carboxylated, one molecule of reduced vitamin K is oxidized (converted to the epoxide form)
Vitamin K epoxide reductase is required to convert the epoxide form of vitamin K back to the reduced form so it can be used again.
Warfarin inhibits the activity of vitamin K epoxide reductase and prevents recycling of the vitamin K back to the reduced form
When Vitamin K cannot be recycled, it is unavailable to the reaction that carboxylates glutamic acid
If reduced Vitamin K is unavailable at the time of zymogen synthesis, the resulting factor is unable to bind Ca++, and thus it cannot bind to cell membranes.
The plasma will contain a heterogeneous population of undercarboxylated forms of the Vitamin K dependent factors.
The undercarboxylated forms have reduced activity
Since warfarin blocks the reductase but not the carboxylase, and prevents recycling of Vitamin K, it is an effective anticoagulant.
Warfarin is often used to treat thrombosis, including pulmonary embolus and deep venous thrombosis
Note that because warfarin only effects the reductase, the effects of warfarin poisoning can be reversed by administration of vitamin K
Nevertheless the dose must be carefully monitored because too much warfarin is dangerous.
A word about Factor XI:
Although Factor XI activates Factor IX in the intrinsic coagulation cascade, a severe factor XI deficiency does not result in a hemorrhagic disorder comparable to VIII or IX deficiency.
XIa (activated by thrombin) associates with platelets on the surface to activate more IX to IXa
Factor IX can also be activated by the TF/VIIa complex on the tissue bearing cells
Factor XI is a only a “booster” of tenase complex formation
Once a clot is formed, the clotting process must eventually terminate
If not turned off, clotting would occur throughout entire vascular tree
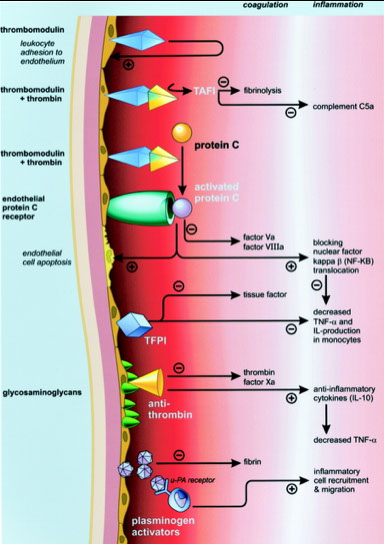
The endothelium is essential in confining coagulation reactions to the site of injury, and preventing clot extension
The non-thrombogenic intima of endothelial cells encourages fibrinolysis and inhibits platelet aggregation through chemical mediators
Protein C/Protein S/Thrombomodulin system
- thrombin formed during coagulation may diffuse or be swept away from the site of injury
When thrombin encounters an intact endothelium, it binds with high affinity to thrombomodulin on the endothelial surface
Thrombin-TM complex activates protein C, which binds cofactor protein S and inactivates Factors Va and VIIIa
Thrombin outside the immediate area of injury is self-inhibiting.
Protease inhibitors
The protease inhibitors AT and TFPI are always present on the surface of endothelial cells , where they can inactivate proteases near an intact endothelium
This serves to further limit coagulation to the site of injury only.
In the next section, we will look at these protease inhibitors more closely.



Fibrinolysis & Anticoagulation
Normal hemostasis is essential in maintaining a healthy circulation. The goal of hemostasis is to:
Minimize blood loss through coagulation
Inhibit and/or remove unnecessary thrombi
Activation of coagulation also activates fibrinolytic activity
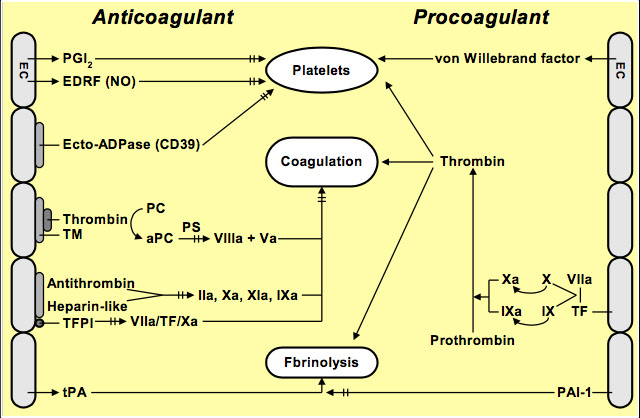
As we have seen, the endothelium plays both a procoagulant role and an anticoagulant role. We will now examine the latter in more detail.
The endothelium exerts three kinds of anticoagulant control:
1) Control of platelets
2) Control of the coagulation cascade
3) Fibrinolysis
Control of platelet function
Prostacyclin (PGI2) - one of most potent physiologic inhibitors of platelets ; also a strong vasodilator
- Its biosynthetic pathway is similar to thromboxane (TXA2), a potent procoagulant platelet agonist
- synthetic paths diverge after the prostaglandin stage
Key point to remember:
PGl2 is produced by endothelial cells
TXA2 is produced by platelets
Control of platelet function
Production of both begins when platelet agonists stimulate the conversion of membrane phospholipid to Arachidonic Acid
At the prostaglandin stage, the pathways diverge because of the action of different enzymes
Prostacyclin synthase for PGI2
Thromboxane synthase for TXA2
Aspirin blocks both pathways at the cyclo-oxygenase stage
The binding is irreversible
Aspirin is used in cardiac disease to prevent thrombosis by blocking the conversion of arachidonic acid to prostaglandin.
Question: How can aspirin exert a net anticoagulant effect if it also blocks production of the platelet antagonist prostacyclin
Thromboxane is produced by megakaryocytes, and is a component of platelets
Platelets have no nuclei
Anucleate cells can’t produce more TXA2
Prostacyclin is produced in endothelial cells
Endothelial can make new cyclo-oxygenase within a few hours to replace the ASA-bound cyclo-oxygenase.
In addition to inhibiting coagulation, endothelial cells secrete chemical mediators which activate fibrinolysis
Tissue Plasminogen Activator (tPA) is released by endothelial cells
It activates plasminogen to become plasmin, which breaks down fibrin clots.
PAI (Plasminogen Activator Inhibitor) circulates and reversibly inhibits the action of tPA.
The endothelium secretes substances which cause nearby platelets to be unresponsive. These include:
Prostacyclin (PG I2)
Inhibits platelet activation, on its own and with nitrous oxide.
Nitrous Oxide
PG I2 and NO act synergistically to inhibit platelet activation.
Both are strong vasodilators
CD39
CD 39 inactivates ADP, which facilitates platelet aggregation
Nitric oxide (NO) is also called Endothelium Derived Relaxing Factor (EDRF)
NO Inhibits platelet adhesion, activation and aggregation
It is also a powerful vasodilator with a short half-life
Nitric Oxide has several effects on the platelets and vascular system involving complex interactions with cGMP, cAMP, and PGL2.
One effect of NO is to decrease intracellular calcium in platelets and smooth muscle. Decreased intracellular calcium results in:
Inhibition of platelet aggregation
Vasodilation in smooth muscle
Working synergistically with Prostacyclin, Nitric Oxide also has the following effects:
Suppresses platelet gpIIb/IIIa receptor
Inhibits platelet conformational changes
Deactivates thromboxane receptors
Modifies proteins critical to platelet activation
CD 39
CD39 is a plasma membrane-bound enzyme that hydrolyzes the platelet agonist ADP to AMP
ADP is necessary for platelet aggregation
This returns platelets to their resting state
Control of the coagulation cascade may occur in two ways:
Swiftly flowing blood disperses activated factors not incorporated into platelet plug
There is an increased risk of thrombosis with blood stasis
Activated factors are then dismantled in the liver
Three regulatory pathways exerted by the endothelium:
Thrombin/Thrombomodulin/Protein C/S complex
Anti-thrombin
TFPI
Thrombomodulin is an endothelial cell integral protein expressed on its surface and given the designation CD141.
Thrombomodulin is a receptor for thrombin, which converts fibrinogen to fibrin, the end product of coagulation.
However, when bound to thrombomodulin, thrombin is inactivated, and fibrinogen cannot be converted to fibrin
Protein C-ProteinS/Thrombomodulin anticoagulant pathway
In addition to being inactivated, thrombin takes on an anticoagulant effect when bound to thrombomodulin.
The complex formed activates protein C, which binds cofactor protein S
The activated protein C/S complex degrades activated cofactors Va & VIIIa
Protein S may be inactivated by C4b binding protein
Protein S is a Vitamin K dependent glycoprotein
It is synthesized by endothelial cells and megakaryocytes
When activated, it serves to limit the coagulation process to the site of injury through its formation of an activated complex with cofactor protein C
Serine Protease Inhibitors
The serine protease inhibitors Antithrombin and Tissue Factor Pathway Inhibitor are always present on the surface of endothelial cells where they can inactivate proteases near an intact endothelium
Antithrombin
Inactivates Thrombin, Factor VIIa, Factor Xa, Factor IXa and Factor XIa (all serine proteases)
It is activated by Heparan sulfate or heparin-like molecule which is anchored to the endothelium of the vessel wall.
Heparan sulfate causes a conformational change in AT
This increases its affinity to activated factors (the serine proteases)
Antithrombin forms a 1:1 irreversible complex with activated factors, causing them to be cleared by the liver.
TFPI (Tissue Factor Pathway Inhibitor) circulates and associates with a lipoprotein.
This association blocks coagulation by binding with activated coagulation factors Xa and IXa, which reversibly inhibits their activity.
When TFPI combines with either Factor Xa or IXa, the resulting complex also inhibits activated factor VIIa
Tissue Plasminogen Activator (tPA)
tPA is secreted from the endothelium
It circulates with its inactivator, plasminogen activator inhibitor-1 (PAI-1), to which it has a strong affinity
The avidity of tPA for fibrin is greater than for PAI-1, so it will let go of PAI and bind to fibrin if fibrin is nearby
The tPA/fibrin site becomes a binding site for plasminogen (PLG) from plasma
Binding to this site activates plasminogen to plasmin, which breaks down the fibrin clot
Plasminogen is converted to plasmin directly on the surface of a fibrin clot
Because it is bound to tPA, the activity of Plasmin is localized to the site of the clot
Any escaping Plasmin is neutralized by α-2-antiplasmin or
α- 2-macroglobulin
Thus plasmin activation is localized to the site of the clot
Plasmin Mechanism
Plasmin acts by cleaving the link between the D and E regions of fibrin
Plasmin can act on both fibrinogen and cross-linked fibrin
If there are no cross-links, it cleaves between D and E regions, leaving D fragments and E fragments.
If fibrin is cross-linked, an additional fibrin degradation product, the D-dimer, is present
D-dimer, a common laboratory test, indicates plasmin activity on cross-linked fibrin
The absence of d-dimers in the presence of fibrin degradation products indicates fibrinogenolysis
Plasmin can act on both fibrinogen and cross-linked fibrin.
Cross-linking of fibrin causes stable links between D moieties
Plasmin cleaves between the D and E moieties.
The breakdown of cross-linked fibrin results in d-dimers
The breakdown of fibrinogen results in fibrin degradation products (FDP), but no d-dimers.
The D-dimer test is used in the diagnosis of thrombosis
The D-dimer in combination with FDP is used to diagnose primary fibrinogenolysis
Lab Evaluation of Hemostasis
To make a proper diagnosis of a bleeding disorder, it is desirable to obtain the following:
- Documentation of the site of bleeding; physical appearance, severity, frequency of bleeding episodes
- Reliable pt and family history of bleeding episodes
- Accurate drug history information
- Presence of any underlying disease
The choice of screening tests for evaluation of a bleeding patient is based on the type of bleeding seen:
- Disorders of primary hemostasis involve vascular tissue and platelets
-- The type of bleeding seen in primary disorders:
Petechiae (pin-point bleeding under the skin)
Epistaxis (nosebleeds)
Superficial &/or mucosal bleeding
Disorders of secondary hemostasis involve coagulation factors, and manifest in a failure to form a stable clot.
The type of bleeding seen in secondary disorders:
- Deep hematomas
- Hemarthrosis
- Delayed bleeding; oozing from a surgical wound
- Delayed healing
Routine screening tests serve to narrow the search for the root cause of the bleeding disorder.
- Primary hemostasis screening tests:
Platelet count (in CBC)
Peripheral blood smear evaluation
Bleeding time or Platelet Function Assay (PFA-100)
- Secondary hemostasis screening tests:
Prothrombin Time (PT)
Activated Partial Thromboplastin Time (aPTT)
Thrombin Time (less commonly done)
Some disease processes may involve primary, secondary, and tertiary hemostasis.
Primary Hemostasis Screening tests include:
Platelet count ; Smear exam ; Bleeding time ; Closure time (PFA)
- Specialized testing (if screening tests indicate platelet dysfunction) : Platelet Aggregation
The Platelet Count is a routine part of a Complete Blood Count (CBC)
- Normal platelets >100K= no symptoms
Life threatening platelet count: <10K
A platelet count will only give information about the number, not the functional competence, of the platelets.
Sources of error: Clotted specimen ; Spontaneous clumping of platelets in EDTA ; RBC fragments
- An examination of the smear must be done to confirm the number of platelets, check for platelet clumps, and look for the presence of rbc frags when the count is in a critical range
Major Causes of Thrombocytopenia
- Decreased production of platelets/megakaryocytes
- Increased consumption of platelets
- Splenomegaly and dilutional effect (overhydration) must first be ruled out before investigating thrombocytopenia.
-- Approximately 30% of platelets are sequestered in the spleen for fast release if needed.
-- If the spleen grows too large, it will sequester more circulating platelets, possibly resulting in tbcpenia
Bleeding Time
The bleeding time is a test of platelet function, capillary fragility, and vascular integrity
- has not achieved acceptable reproducibility (not an effective screening test)
- can be done to screen for qualitative plt disorders and vWD, though the PFA is more sensitive
- done at the patient’s bedside (not done too much)
- cuff is used to apply standard pressure (40 mmHg)
- small incision is made on the patient’s inner forearm
- A stopwatch is started at the time of incision
- The wound is blotted at 30 second intervals
The stopwatch is stopped when bleeding has stopped
The Bleeding Time tests for:
Platelet competence and Vascular integrity
Sources of error:
Standardization of the cut is difficult
Platelet plug may be pulled off when blotting
Results are not reproducible
Closure time (Platelet Function Assay, PFA-100)
Measures time to close standardized aperture (closure time), c shear forces simulating a BV, with the aperture covered by either Collagen and epi (Col/epi; sensitive to global plt defects) or Col/ADP (good for drug-induced plt defects)
- standardized & accurate measure of plt function
- Blood passes through an aperture coated with collagen (which stimulates adhesion) + ADP or epinephrine (which stimulate aggregation and activation)
The time it takes for the platelets to form a plug sufficient to close the aperture is measured
- PFA tests plt function only, not vascular integrity; but is also affected by plt number and hematocrit
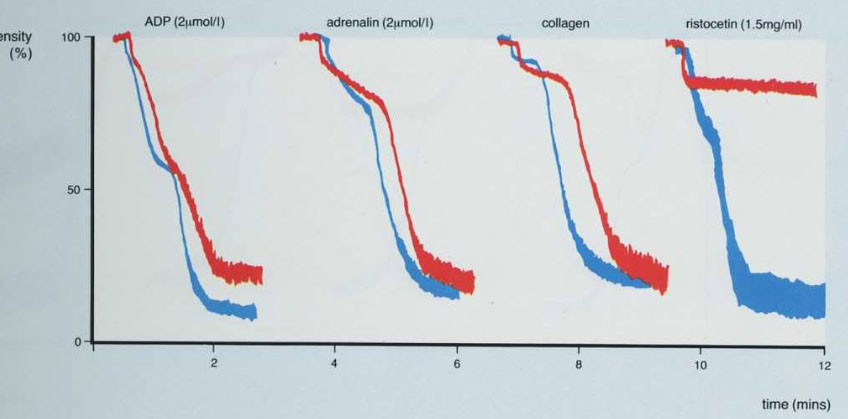
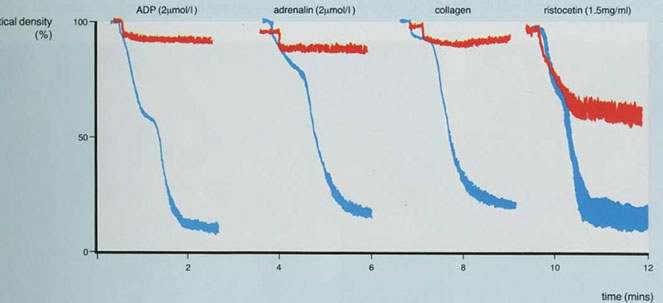
Platelet Aggregation studies (Platelet aggregometry)
Done on plt rich plasma (slowly centrifuged whole blood), where it is stirred continuously in cuvette and exposed to agonists (ADP, epi, collagen, ristocetin)
- not a routine test, but is done when abnormalities are revealed by the PFA.
- Platelet aggregation studies measures aggregation of platelets in the presence of platelet agonists
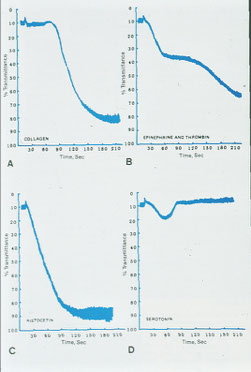
Aggregation of platelets is measured by photo-optical means - Aggregated platelets cause a decrease in light transmittance through platelet-rich plasma soln
- Different platelet disorders have characteristic aggregation patterns
Biphasic curve c low dose ADP and epi (second wave 2/2 plt degranulation)
- monophasic curve (primary aggregation only) c high dose ADP, Collagen (usually c a short lag time) and Ristocetin (response to ristocetin seen over 1.2 mg/mL [Ristocetin])
- firefly luminescence assay can measure release of platelet dense granules in secondary wave
Abnormal aggregometry usually 2/2 rx (aspirin), whereas a poor response to all agents except ristocetin seen Glanzmann tbcpenia (rare; so Glanzmann only responds to ristocetin)
- storage pool defects and aspirin can cause absence of secondary phase c ADP and epi
- MPNs can cause poor response to epi as well
- vWD and Bernard-Soulier dz shows response to everything except ristocetin
Sources of error: Lipemia (interferes with optical readings) ; Hemolysis (intact RBCs contain ADP) ; Elapsed time (must be tested within 3 hours of collection)
- thus is highly sensitive to preanalytic conditions
Defects in Secondary hemostasis may be due to:
Genetic abnormalities
Hemophilia A- Factor VIII
Hemophilia B- Factor IX “Christmas Disease”
Other factor deficiencies rarely have a genetic cause
Von Willebrand Factor Deficiency (may appear to be a primary defect)
- Acquired abnormalities: Medications; Vitamin K antagonists; Vitamin K deficiency; Liver disease
- involve tests which evaluate the formation of fibrin.
Including tests of:
Intrinsic pway; Extrinsic pathway ; Common pathway
Tests of secondary hemostasis may be either functional or quantitative.
- Functional Assays detect formation of a fibrin clot by the following means:
Optical (most common); Electro-Mechanical (older method); Viscosity (magnetic); Chromogenic
- Quantitative Assays detect the presence of individual coag factors
Quantity may be normal, but function may be abnormal
- Quantitative assays are done by the following methodologies:
Immunoassay; Electrophoresis
Pre-analytic errors effecting tests for secondary hemostasis
- Reliable test results can only be obtained from adequate test specimens. Following are possible sources or pre-analytic errors:
- Problems with blue-top tube: Partially filled tubes, Vacuum leak and citrate evaporation
- Problems with phlebotomy: Misidentification of patient; Heparin contamination (blood drawn from a picc line without flushing); Slow fill; Underfill; Vigorous shaking
- Biological effects: Hematocrit ≥55 or ≤15; Lipemia, hyperbilirubinemia, hemolysis (optical methods)
- Laboratory errors: Delays in testing (> 2 hr post draw); Prolonged incubation at 37°C; Freeze/thaw deterioration
Activated Clotting Time (ACT)
Usually done as POC to monitor high-dose heparin tx that is often s/p cardiopulmonary bypass surgery
- done on whole blood collected in tube containing intrinsic pathway activator (kaolin, glass) and analyzer measures clotting time
- not reliable as aPTT or anti-Xa assay bc inflienced by lots of non-coag parameters (hypothermia, hemodilution, plt defects)
The Activated Partial Thrombin Time (aPTT)
Measures coagulation factors in the intrinsic and common pathways.
Thrombin Time (TT)
Thrombin added to pt plasma, the time to clot formation is the thrombin time
- TT prolongation caused by deficiency or defective fibrinogen or inhibitors (heparin, hirudin, argatroban, FDPs)
- can confirm presence of heparin c prolonged TT if reptilase time is normal and 1:1 mix fails to correct
Reptilase time
Take Bothrops atrox venom, add PPP, measure clot time
- inc reptilase time in dysfibrinogenemia
If pt on heparin, will have in inc TT but normal Reptilase time
- Reptilase measures conversion of fibrinogen to fibrin, but is insensitive to heparin
- heparinase and protamine will correct TT that is inc in a pt on heparin
Prothrombin Time (PT)
Measures coagulation factors in the extrinsic and common pathways (may be more sensitive than aPTT for measuring common pathway deficiencies)
Coagulation factors measured: I, II, V, VII, X
Principle: Excess calcium is added to citrated plasma incubated at 37◦
1. This reverses the effect of citrate and allows coag factors to become activated
2. Tissue thromboplastin is added to the solution to activate the extrinsic pathway
3. Time (in seconds) is measured until a clot forms.
Use of the protime in monitoring anticoagulant therapy to monitor Coumadin (Warfarin) therapy
- Coumadin (Warfarin) is a vitamin K antagonist.
- Factors II, VII, IX, and X are dependent on vitamin K for proper function
- speed of the extrinsic pathway is greatly affected by the amount of functional Factor VII available
- Factor VII has the shortest half-life of the vitamin K dependent coagulation factors
- decrease in Factor VII activity detected below 50%
- Decreases in other extrinsic pathway factors will be detected below 30% activity
INR- International Normalized Ratio
- result (in seconds) of a protime will vary depending on the Tissue Factor reagent used.
- Different batches of TF will vary between manufacturers, and between lots from the same manufacturer.
- INR used to standardize results between labs
- Manufacturers compare each batch of TF with an International Standard, and assign them an International Standard Index (ISI) value
INR= (patient PT (sec)/control PT (sec))^ISI
ISI = ISI reference preparation x regression line slope
- when trying to establish the control value, use the geometric mean of a given population
INR used to adjust patient's drug dosage to get PT into desired range, right for patient and their dx
- For most patients, the therapeutic range is typically between 2.0 and 3.0 INR, but may be higher in patients who tend to form thrombi.
- INR not used in evaluation of normal patients, patients with liver disease, or patients transitioning from heparin to coumadin therapy
Activated Partial Thromboplastin Time (aPTT)
Phospholipid (contact activator of fXII [silica, kaolin) and excess Ca2+ added to citrated plasma, where the time to clot formation is the aPTT
- prolongation can be 2/2 factor deficiency or inhibitor
-- used to monitor heparin tx (unfractionated heparin, where the therapeutic range is 1.5-2.5x the control)
Coagulation factors measured: I, II, V, VIII, X, IX, XI, XII
Principle:
1. Platelet poor plasma is incubated at 37◦
2. Phospholipids and a contact activator (e.g. Kaolin, micronized silica or ellagic acid) are added, followed by calcium
3. Time (in seconds) is measured until a clot forms.
The aPTT is used to monitor heparin therapy
Inpatient only
The aPTT will be prolonged in patients on long-term coumadin therapy, but the PT is a more sensitive test for coumadin therapy.
- elevated fVIII can shorten aPTT
If aPTT is prolonged, mixing study is done
- mixing c different blood products can help determine the missing factor (aged plasma lacks fII, V, VIII, XIII and serum lacks fibrinogen and fII, V, VIII, XIII)
- if mixing study corrects aPTT factor deficiency suspected
- if mixing study does not correct aPTT, then an inhibitor is suspected
Pretest variables can mess with the results
- high hematocrit has less plasma, which messes with the ratio of plasma to anticoagulant (trisodium citrate) and can falsely prolong aPTT, with a similar result seen in tubes that are underfilled (overfilling has opposite effect)
- photo-optical methods can be messed up if sample is lipemic, icteric, turbid or hemolyzed
- plasma should be separated from cells ASAP bc plts release PF4, shortening PTT
- neonates have longer aPTTs, which shorten at ~6 mo age
Heparin Antifactor Xa assay (Anti Xa assay)
Used to monitor either unfractionated or low molecular weight heparin
- pt plasma added to known amt of fXa c excess antithrombin
- heparin in pt plasma stimulated antithrombin to inhibit fXa, and the quantity of residual fXa measured c chromogenic substrate, with the residual Xa subtracted from initial Xa to determine anticoagulant concentration
Sample taken 4 hours after dose of LMWH, and testing done within 1 hour
- the test is not affected by underfilled tubes (like the aPTT is) and is not affected by abnormal factor levels
Factor VIII inhibitor assay and Factor VIII antibody assay (Bethesda assay)
- done to find the titers of anti-fVIII ab
- done by making dilutions of pt plasma c citrated saline, mixing 1:1 with normal plasma after each diltuion and incubating for several hours
- Factor VIII assays done on each dilution, and the dilution where fVIII is 50% is the inhibitor titer, with results expressed in Bethesda units
- low ab titers, <5 Bethesda units, can be tx'd c DDAVP and higher doses of human fVIII
D-dimer and Fibrin Degradation Products (FDP) / Fibrin Split Products (FSP)
fXIII makes D-dimers when D regions of fibrin molecules covalently linked
- fibrin clots dissolved by plasmin, which is incapable of breaking up D-dimers
- presence of D-dimers indicates that fibrin has been formed and then degraded
- tests for FSP is less specific bc detects fibrin degradation and fibrin degradation products
- D-dimers and FDP/FSP inc in thrombosis and significant bleeding (can also be inc in atrial fibrillation, CHF and cirrhosis)
- high sensitivity D-dimer is D-dimer assay approved by FDA for exclusion of DVT/PE
Factor Assays
- can measure fII, V, VII, VIII, IX, X, XI, XII by mixing pt plasma c reagent plasma c known factor deficiency and then comparing the resulting PT and PTT against a standard, usually by performing serial dilutions
- inhibitor suggested if serial dilutions cause inc in activity of factor
- normal plasma has 100% activity by definition (1 U/mL) for each factor, though in reality adults have activity levels bwt 60-150% normally
Fibrinogen activity
the MC fibrinogen assays (Clauss and Ellis assays) are similar to thrombin times
- lower fibrinogen levels = longer clotting times
Fibrinolysis
- no “routine” tests for fibrinolysis.
- Tests for fibrinolysis are done to investigate whether a patient has a thromboembolus
- Fibrin Split (or Degradation) Products are result of breakdown of fibrinogen or fibrin, whether or not it has been cross-linked.
- D-dimers are breakdown products of fibrin which has been cross-linked
Elevated levels of D-dimer are almost always present in cases of venous thromboembolism.
However, elevated levels are seen in a number of other conditions seen in hospitalized patients
Therefore, a positive result is not indicative of VTE.
A negative result can rule out VTE with about 90% certainty
FSP and D-dimer tests are used as an aid in diagnosing Disseminated Intravascular Coagulation (DIC); however, a discussion of this complex disease process is beyond the scope of this lesson
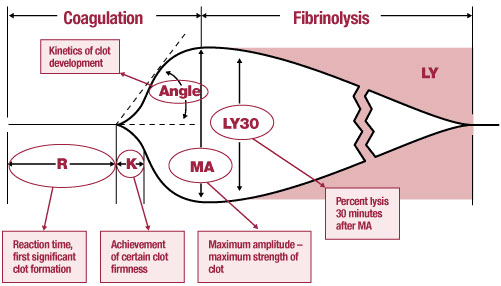
Thromboelastogram
Measures the global visco-elastic properties of whole blood clot formation
- if R inc = give FFP
- if K inc = give cryo

Closure time tests for platelet aggregation when exposed to collagen in the presence of ADP or Epineph
- often used to diagnose aspirin resistance, and is a good screen for von Willebrands Disease.
Sources of error:
Hematocrit < 28 ; Platelet count < 100
Platelet Aggregation study
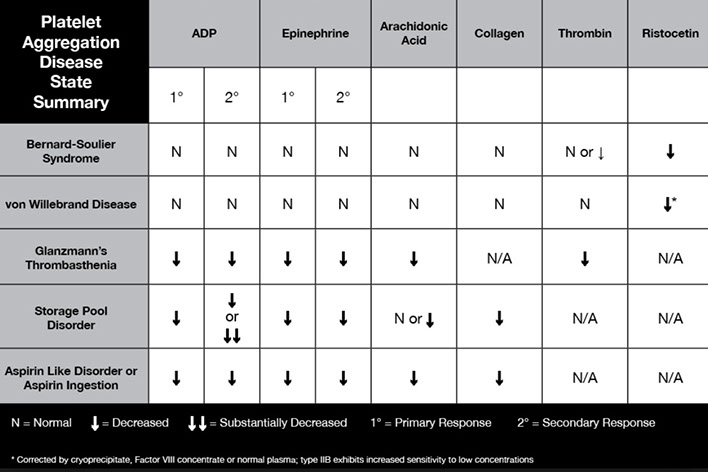
Normally, there is aggregation with all agonists except low-dose ristocetin
vWD IIb and pseudo-vWD (platelet type) show aggregation with low-dose ristocetin


Hemophilia
- causes of excessive bleeding
Pts c plt-type bleeding have easy bruising, petechia and purpura 2/2 Tbcpenia, plt malfunction and vWD
- aqd dz like ITP, EtOH intox, aspirin, penicillin, uremia, DIC and MPNs as well
Pts c coagulation-type bleeding have hemarthrosis, intramuscular hematoma 2/2 coag factor disorder / inhibitor or vWD
Unexpected Tbcpenia should be confirmed c PBS to exclude: plt aggs and satellitosis (which cause pseudo-Tbcpenia), markedly large plts (can look bigger c ITP), schistocytes
Giant plt disorders:
Bernard-Soulier syndrome (BSS)
Mediterranean macrotbcpenia (mild BSS)
May-Hegglin anomaly (giant plts c Dohle bodies)
Fechtner syndrome (giant plts c hearing loss, cataracts and nephritis)
Sebastian syndrome (giant plts c leukocyte inclusions)
Gray plt syndrome
ITP
Montreal plt syndrome
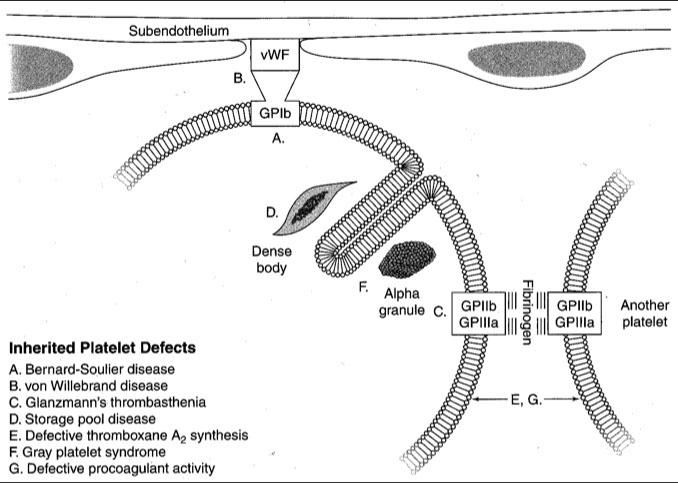
Bernard-Soulier Syndrome (BSS)
*** plts are as big as St Bernard!!! ***
- 2/2 AR trait causing dec / defective Gp1b / V / IX complex (vWF receptor) causing giant plts and tbcpenia
- aggregometry shows aggregation c all agonists except ristocetin (similar pattern as vWD), except in BSS there is failure to agg even c normal plasma (vs vWD which aggs in normal plasma)
- flow cytometry on plts shows dec Gp1b / V / IX (CD42b)
- carrier state is Mediterranean tbcpenia
Plt Storage Pool Dz's
- includes abnormal dense granules (Hermansky-Pudlak, Chediak-Higashi, and Wiskott-Aldrich) and abnormal alpha granules (gray plt syndrome)
- dec plt agg 2/2 def in either dense granules / alpha granule content or both
- normal morphology, no granules on EM
- plt agg studies show NO 2nd wave-ADP, EPI
- dec collagen, +AA, normal ristocetin
- inc ATP:ADP ratio
Dense granule disorders
- only 1st wave agg seen, ATP secretion c chemiluminescence markedly diminished and no dense granules on EM
Hermansky-Pudlak syndrome
- common in Puerto Rico (PR), presenting as epistaxis in kiddos c oculocutaneous albinism
- can get complicated by pulmonary fibrosis and granulomatous colitis
- bx shows ceroid-like material in alveolar macrophages
- genes HPS1-6, AP3B1 and DTNBP1
Chediak-Higashi syndrome
- ceroid bodies in PMNs, plts, and melanocytes
- oculocutaneous albinism complicated by lymphoproliferative dz's
Wiskott-Aldrich syndrome
X-linked disorder c triad of tbcpenia, eczema and immunodef from WASP gene defect
Platelet alpha granule disorders
Gray platelet syndrome
- recessively inherited mild bleeding disorder where agg is blunted c all agents except ADP
- lack of plt alpha granules causes uniform pale gray plts on Wright-stained PBS
- plt aggregation studies are generally normal
Glanzmann thrombasthenia
Deficient GpIIb/IIIa complex
- GpIIb is fibrinogen receptor and GpIIb/IIIa complex is PLA1 ag, so plts are PLA1 negative
- aggregometry shows agg failure c all agonists except ristocetin, the opposite of BSS and vWD
- see diminished clot retraction on in vitro clot retraction test
- flow shows dec GpIIb/IIIa (CD41/61)
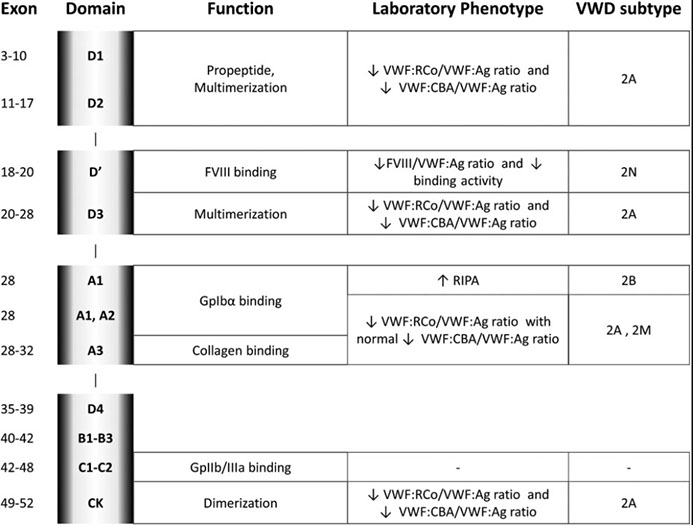
von Willebrand Disease
- MC inherited bleeding diathesis (hemophilia A is MCC of severe hemophilia)
- impaired coagulation cascade (vWF maintains fVIII) and plt function (helps form plt thrombi)
- vWF normally formed in endothelial cells (in Weber-Palade bodies) and Mgkcs (in alpha granules)
- secreted by endothelial cells into BM, which binds plts when exposed by activation of Gp1b
- if secreted into circulation is broken down by ADAMTS13 into dimers and multimers (seen on electrophoresis)
- OCPs can elevate vWF and lead to delayed dx
- several different types:
vWD type I (7/10) is mild quant defect c nirmal PT, plt count long PTT, BT and PFA-100 CT 2/2 dec fVIII, vWF ag, and vWF activity (called "concordant" bc vWF activity to vWF ag ratio is ~1)
- only type tx'd c DDAVP
vWD type IIa (3/20) is missense mut in vWF gene exon 28 c abnormal multimer patter (absent high molecular weight multimers) 2/2 markedly dec vWF activity and normal to low fVIII and vWF ag causing increased susceptibility to ADAMTS13 proteolysis and thus mod to severe bleeding c long PTT and "discordance" (vWF ag : vWF activity < 0.7; markedly decreased ristocetin aggregation)
vWD type IIb has dec large multimers c enhanced ristocetin induced plt agg (plts are stickier than normal) and profound tbcpenia and bleeding when exposed to DDAVP 2/2 gain-of-function mut of GP1b causing spontaneous binding of abnormal vWF to normal plts; aggregates with low levels of ristocetin, whereas controls do not; this type must be distinguished from plt type vWF (very similar) done with cryoprecipitate challenge (which has fVIII and causes ; treated with vWF / FVIII
*** 2D's = B ***
vWD type IIM is loss of function mutation in Gp1b binding domain that prevents binding of vWF to Gp1b, though the multimer analysis is normal
*** the Mut (like St Bernard Dog) Mutation ***
vWD type IIN (Normandy) has mutation of the domain binding fVIII causing low levels of circulating fVIII mimicking hemophilia A, with vWF quantity and function otherwise normal
- can look like hemophilia a, usually in women
*** D-Day (stormed the beaches of Normandy) was in 1944 (4+4 = 8! the factor deficient here ) ***
vWD type III is severe with virtually no vWF, low fVIII and faint multimers, 2/2 various muts in vWF gene
- decreased Weibel-Palade bodies in endothelial cells
pseudo- / plt-type (PT) vWD has the lab findings of vWD type IIb, but is caused by abnormal Gp1b-alpha causing inc avidity for vWF; aggregates with low levels of ristocetin, whereas controls do not;
- mutations in GP1a causing inc binding of GP1ba to HMW vWF multimers
- AD inheritance
- vWD can be acquired 2/2 lymphoprolif dz, autoimmune dz, ET, or aortic stenosis
- grp O blood pts have a little less vWD than other blood groups, though levels can be raised by exercise, inflam, preg and E
- newborns have high vWF and disorders might not be apparent until age 1 yr
- treated with platelet transfusions
Labs: gold standard for classification is vWF multimer analysis on gel electrophoresis
- can monitor dz during surgery using fVIII activity
- ristocetin cofactor assay (vWF activity assay) done by adding pt plasma to formalin fixed normal plts c ristocetin and measuring agg (will not fully agg in vWD)
- can measure vWF: AG by ELISA or immunoturbidometric assay
- Plt aggregometry (discussed above)
Hemophilia A
- MC inherited severe hemophilia, XLR, though family hx absent in up to 1/3 pts (have new spontaneous mutation)
- classify severity 2/2 amt of bleeding: severe if spontaneous bleeding occurs (fVIII <1U/dL), moderate if excessive bleeding c minor trauma (fVIII 1-5 U/dL), or mild if excessive bleeding c surgery or major trauma (>5 U/dL)
Labs: normal plts, PT and TT, PTT elevated and completely corrects, isolated fVIII def (should also consider vWD once fVIII def discovered)
- female carriers have >50% fVIII activity, normal PTT, no hx of bleeding, but abnormal fVIII to vWF ratio of ~1:2 (normally 1:1)
- factor VIII gene on Xq28 is large c 26 exons, allowing for lots of possible mutations (usually 2/2 intron 22 inversion in ~1/2 of affected whites)
- 1/3 of pts getting fVIII replacement tx get fVIII abs, which can be confirmed c Bethesda inhibitor assay
-- also can happen in female neonates and the elderly c severe bleeding, will see isolated elevated aPTT that doesn't correct in mixing study and confirmed c Bethesda assay
Tx: recombinant FVIII; maintain at >1%
- in case of head trauma c intracranial bleed need to give 100% fVIII replacement
Bethesda unit (BU): vol of plasma that inactivates 50% of fVIII activity in normal plasma
- BU determines the strength of an antibody present; if <5 BU then can attempt to overwhelm inhibitor with excess replacement factor, but if >5 BU then need to use alternating concentrates (porcine factor VIII) or bipass agents (factor VIII inhibiting bypass activity, prothrombin complex concentrates, recombinant f VIIa) used to control active bleeds
dose of factor VIII = % inc factor x kg x 0.5 IU/kg
- goal is 50% with dental extraction, hemarthrosis, GI bleeds
- goal 100% for intracranial bleeds
- t1/2 of fVIII is 12 hrs, so maintenance dose (1/2 of the loading dose) given every 12 hrs
Hemophilia B (Christmas dz)
- XR congenital deficiency of fIX, thus inc PTT (can be normal) that corrects c mixing study and normal plts, PT and TT
- gotta exclude vit K def (liver dz, fat malabsorption, coumadin use)
- fIX Leiden is hemophilia B c spontaneous remission after puberty
Isolated factor deficiency (II, V, VII, X. XI)
- AR, c homozygotes having 1/20 the normal activity, but not as severe as hemophilia A/B
- MC is fVII def, with amt of bleeding not correlating to fVII levels, and inc PT of course
-- thrombosis is a rare complication of fVII def assoc c surgery and fVII recombinant tx
-- recombinant fVIIa has a short T1/2 and has to be given every 2 hours
- fV def c characteristic PT and PTT prolongation, normal TT
- fX def c PT, PTT and DRVVT inc, normal TT; fX def also assoc c amyloidosis (binds directly to fX)
- fXI def in Ashkenazi Jews, in up to 1/10, usually mild, and is seen in Noonan syndrome
To calculate the dose of factor to be given, multiply:
% activity desired x Body Weight x [Dose] (in IU/kg)
Factor XIII deficiency
- also AR, mild bleeding in heterozygotes, c umbilical stump bleeding, recurrent miscarriages, characteristic delayed bleeding, hypertrophic scars
- if homozygous, PT, PTT and TT normal, but pt has severe bleeding, which can be life-threatening at birth in umbilical stump and intracranially; should test c clot stability test
- has characteristic rapid clot dissolution in 5M urea or 1% monochloracetic acid
Inherited Combined Factor Deficiency
- 2/2 mutations in intracellular transport or posttranslational modification of multiple clotting factors
- MC is fV and fVIII def (type I) c LMAN1 gene mutation
Afibrogenemia, Hypofibrinogenemia, Dysfibrinogenemia
- can be congenital or aqd, quantitative or qualitative
- in inherited afibrinogenemia and hypofibrinogenemia have super prolonged PT and PTT c bleeding similar to hemophilia A manifesting as umbilical stump bleeding
- 1/2 of dysfibrinogenemia are asx, others can cause bleeding or thrombosis, depending on the mutation
- dysfibrinogenemia causes prolongation of thrombin time, except in Olso I and Valhalla types
- dysfibrinogenemia is MC 2/2 liver dz,
Acquired Factor Deficiency
- fX def seen in amyloidosis sometimes
- liver dz caused dec synth of factors, c inc PT, hypo- or dysfibrinogenemia (inc TT), impaired D-dimer clearance
- vit K def (and warfarin tx) causes dec vit K-dependent factors (***1972***) from dec carboxylation, with fVII having the shortest T1/2
- hemorrhagic dz of newborn presents after ~3 days postpartum
- prolonged use of abx can dec vit K absorption
- pesticides can have a warfarin-like affect
Other causes of bleeding include vascular and CT disorders
- CT fragility (elderly, glucocorticoid excess), scurvy, amyloidosis, vasculitis, fat emboli, Ehlers-Danlos, hereditary hemorrhagic telangiectasia, Fabry dz, ataxia telangiectasia
Disseminated Intravascular Coagulation (DIC)
Occurs when substance in the blood behaves like tissue factor and causes widespread microvascular thrombi and consumes factors
- thus see paradoxical inc bleeding and thrombosis
- usually caused by infx (Gram neg), obstetric complications, mucin-secreting adenoca, trauma, prostatic surgery, snake bites
- labs: inc D-dimer (most sens), low plts, low fibrinogen (most spec), inc PT and PTT
- nothing really sensitive or specific, can use the Intl Society of Thrombosis and Hemostasis (ISTH) score
- tx underlying cause
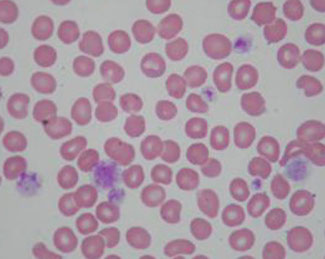
BSS
BSS - big platelets with normal granularity
Gray platelet syndrome - plts without alpha granules
Glanzmann thrombasthenia
vWD


von Willebrand multimer analysis




Neonatal alloimmune thrombocytopenia (NAITP)
Caused by abs specific for plt ags inherited from the father and absent in mom
- in whites, MCC of NAITP is ab against HPA-1a
-- although plts express HLA, abs against HLA rarely cause such severe tbcpenia
- plts also have ABO ags but abs against these cause anemia
- plts do not have HNA or Rh ag
Thrombophilia
- causes of excessive thrombosis
May be able to narrow down ddx by noting where thrombsis occuring (arterial vs venous), but not very specific
- labs should be done 2 mo after acute episode and 2 wks after anticoagulant cessation, though gene testing (Prothrombin 20210 or fV Leiden) can be done at any time
Activated protein C resistance
- aka factor V Leiden, MCC of hereditary thrombophilia, presenting as venous thromboembolism or recurrent miscarriage
- 2/2 AD G to A point mutation at nucleotide 1691 in factor V gene (glutamine for arginine at position 506 in factor V gene) causing resistance of factor V Leiden to APC cleavage
- inc risk c fV R2 (A4070G) mutation or compound heterozygosity (concommitant G20210A mutations, protein C def, AT def)
- activated protein C (APC) normally degrades fV and VIII, thus inhibiting coagulation
- APCR heterozygous in 1/20 whites (less common in other ethnicities)
- Labs: 2 aPTTs done, 1 c pt plasma alone and other in presence of APC, with a clotting time ratio with and without APC (is normally >2)
- see a falsely low protein S on PTT-based protein S activity assay
- should be careful in neonatal dx, as they normally have protein C resistance from low endogenous protein C levels
- can do DNA-based genotypic analysis
Prothrombin Variant (Prothrombin G20210A mutation)
- G to A substituation in prothrombin gene causing inc prothrombin levels
- AD, similar incidence as factor V Leiden (1/20 in general population, 1/5 pts c thrombophilia) and in 1/10 pts c factor V Leiden mutation
- pts present c venous thromboembolism, miscarriage and inc risk of cerebral vein thrombosis
- Labs: usually inc prothrombin levels, but gene sequencing by PCR is preferable
Antithrombin Deficiency
Antithrombin (AT) normally inhibits multiple factors, and when bound to heparin AT activity is enhanced, which is the mechanism of heparin anticoagulation
- AD, pts present c venous TE (risk inc 10x in heteros, homozygous mutations are incompatible with life)
- risk is esp inc in preggos, bc AT normally dec in preg and inc risk of TE in preg
- causes failure to inhibit fIIa (thrombin), Xa and other activated factors
- acqd AT def in nephrotic syndrome, L-asp tx, E, preg, acute thrombosis, DIC and colitis
Labs: if AT <60%, consider AT def if pt >6 mo age
- can have lots of different mutations, MC in SERPINC1 gene for AT
Protein C and Protein S deficiency
Activated Protein C (APC) normally inhibits coagulation by degrading activated fV and VIII, with protein S being the cofactor for protein C
- up to 1/200 incidence in general population, causing up to 1/20 cases of thrombophilia
- both are AD, c coumadin-induced skin necrosis (usually in obese middle-aged women; bc of vit K deficiency?), homozygotes c purpura fulminans as neonates, a rapidly fatal form of DIC requiring immed tx c anticoagulation and FFP
- functional (coag-based) and antigenic (immunologic) assays can be done for proteins C and S (functional assays preferred in screening), c protein C levels <55%(careful in dx'ing newborns as is normally pretty low)
MTHFR gene mutation and hyperhomocysteinemia
- severe homocysteinemia can cause spillage into urine (cysteinuria), with mild to mod forms causing atherosclerosis and AV TE
- aqd mild hyperhomocysteinemia seen in elderly, smokers, renal failure, dec folate, B12, B6
- inherited mild to mod hyperhomocysteinemia 2/2 MTHFR gene mutation in the T allele (C677T), aka thermolabile variant, which is quite common and related to atherosclerosis
- may be able to tx c folate or B6 supplementation
- hyperhomocysteinemia causes endothelial cell toxicity and disruption of vascular hemostatic mechanisms
- recommended tx goal is to lower to <10 umMol; can also tx by giving vit B12, vit B6 and folate
Factor XII deficiency
- marked aPTT prolongation (initially corrects c mixing but then prolongs after incubation), see AV thrombosis, not bleeding diathesis
Antiphospholipid (APL) Syndrome
- includes Lupus anticoagulant (LAC) and Anticardiolipin (ACA) syndromes
-- both have assoc c SLE, HIV, malignancy, rx, can be caused by thrombosis and pulmonary HTN, recurrent miscarriage (1st tri) fetal wasting (2nd and 3rd tri) placental vasculopathy and variable maternal tbcpenia and assoc c tbcpenia in general
- ACA more assoc c premature coronary atherosclerosis, livedo reticularis, a specific postpartum syndrome (c fever, chest pain, pleural effusions, patchy lung infiltrates, cardiomyopathym arrhythmias) and warfarin resistance
- LAC more commonly has prolonged PTT (not usually seen c ACA)
Labs: must have either ACA ab or LAC on 2 occasions 2-3 mo apart
- Dilute Russell viper venom test: Russell Viper venom + factor V, phospholipid and Ca2+ activates Factor X; plasma containing lupus abs prolongs PTT
- LAC testing is clot-based on the principle that LAC prolongs phospholipid-dependent clotting time c failure to correct in 1:1 mixing study, though it does correct c excess phospholipid; the aPTT or DRVVT (more sens) can be used
- ACA testing is ELISA based, c high IgG ACA titers; if IgG titers low or have only IgM, pt is at inc risk of getting IgG titers later on
-- ACA test can be false pos in syphilis, Q fever and HIV (usually IgM)
- false + nontreponemal VDRL/RPR
Heparin Induced Thrombocytopenia (HIT)
Type I HIT
- early and mild tbcpenia, c plt count dec on day 1 or 2 of heparin tx, which doesn't dec >1/2 of baseline
- occurs in 1/20 pts on heparin, is not a danger to pt and is not immune-mediated
Type II HIT
- plts drop >1/2 of baseline after 1-2 wks of heparin tx, c very high risk of thrombosis, and is immune- mediated
- can have arterial thrombosis (in extremities, CNS, heart) venous thrombosis (lower limb, lung, upper limb, adrenals), microvascular thrombosis (esp in pts on coumadin), erythematous plaques and necrosis at site of heparin injection
- females post-surgery at esp inc risk
Dx: suspect based on time after initiation of heparin tx, amt of plt dec, presence of thrombosis
Labs: anti-PF4 ab fairly sens and spec (~90%), can confirm c serotonin release assay (SRA) and heparin-induced plt agg assay (HIPA), usually send out tests
- Serotonin Release Assay (SRA) is the gold standard for dx of HIT, used in confirmation
Thrombotic Thrombocytopenic Purpura (TTP)
- can be idiopathic, familial (Upshaw-Shulman syndrome), secondary to preg, HSC, rx
- pentad: tbcpenia, MAHA, neurologic abnormalities, renal probs, fever
- plts <20K and numerous schistocytes on PBS
- serum LDH usually inc > 1000
2/2 abnormally large circulating vWF multimers from defective ADAMTS-13, which normally cleaves multimers into smaller ones
Tx: plt transfusion contraindicated
- should do daily plasmapheresis c FFP replacement
Coumadin-induced skin necrosis in obese middle age women from vit K def in more fatty areas
Therapeutic agents and monitoring
Warfarin (coumadin)
Inhibits posttranslational modification of vit K dependent coag factors: FII, VII, IX, X and protein C and S (1972, C S)
- various factors (age, diet) can alter its effects
- genes VKORC1 (Vit K epOxide Reductase Complex) and CYP2C9 have the greatest effects on coumadin
-- pts c CYP2C9*2 and CYP2C9*3 have impaired metabolism of S-warfarin causing inc plasma concentration of the drug despite standard dosing (pts at inc risk of life-threatining bleeding complications)
- MC monitoring is through PT with INR, with INR bwt 2-3 for most indications
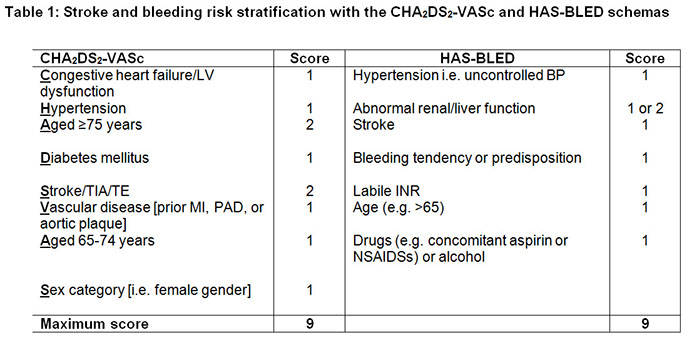
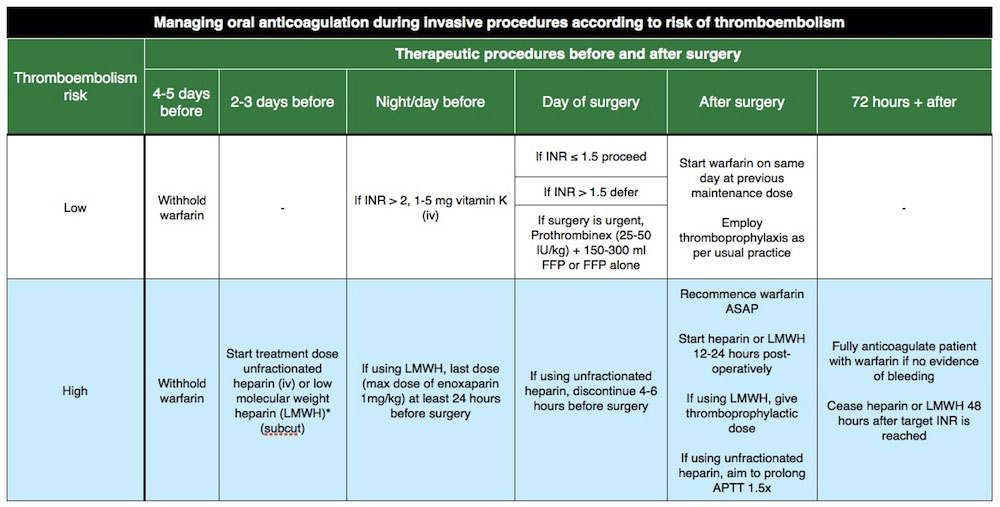
- may need to reverse if INR supratherapeutic or c bleeding (see table)
- purple toe syndrome is a side effect of warfarin occurring 8 wks after initiation of therapy 2/2 cholesterol emboli, and may also cause osteoporosis (like heparin)
Unfractionated Heparin (UH)
Heterogenous mix of mucopolysaccharides ranging in size
- Heparin binds AT, enhancing ATs ability to inactivate thrombin and fXa, IXa, XIa, and XIIa and by activation of heparin cofactor II (HCII, a thrombin inhibitor)
- aPTT best to monitor (calibrated to Xa curve), but can use anti Xa assay as well
- heparin resistance usually 2/2 AT def
- discontinuation of heparin causes normalization of aPTT in 30 min to 1 hr, can use protamine sulfate to rapidly reverse
- can cause osteoblast suppression and activation of osteoblasts leading to osteoporosis
Low molecular weight heparin (LMWH)
- includes enoxaparin, dalteparin, nadroparin, tinzaprin and danaparoid (considered "heparinoid")
- similary, LMWH works c AT to inactivate Xa and thrombin
- monitoring not reqd in all pts, usually just those c severe obesity, preg, renal failure
- monitored using Xa assay, done 4 hrs after injection
- protamine sulfate does not reverse LMWH as well as UH
- can cause osteoblast suppression and activation of osteoblasts leading to osteoporosis, more frequently than UH
Fondaparinux (Arixtra)
- direct Xa inhibitor, that binds AT and causes Xa inactivation, but does not enhance thrombin inactivation
- monitor c aPTT
- no reversal c protamine
Direct thrombin inhibitors (DTI)
- inhibits thrombin w/o AT
- hirudin comes from salivary glands of leeches, recombinant forms are lepirudin, desirudin, argatrobaan and bivalirudin
- argatroban used to prevent thrombosis in pts c HIT
Apixaban and rivaroxaban are direct Xa inhibitors that can be taken PO
- dabigatran is a direct thrombin inhibitor
Aspirin (acetylsalicylic acid)
- anti-plt effects through selective irreversible inhibition of plt cyclooxygenase (COX-1), an in the TXA2 cascade that causes plt activation and vasoconstriction
- pts c atopic disorders can have aspirin allergy
- aspirin resistance seen in 1/4 adults
Thieopyridines
Ticlopidine (Ticlid) and clopidogrel (Plavix) block the P2Y12 receptor, which mediates ADP plt activation
- some pts also can be resistant, usually related to P450 enzymes
Dipyridamole
Plt inhibitor that reduces intracellular Ca2+ via phosphodiesterase inhibition
- called Aggrenox in pill c aspirin
GpIIb/IIIa receptor antagonists
includes abciximab (ReoPro), eptifibatide (Integrilin) and tirofiban (Aggrastat)



COVID 19

D-dimer
References
1. Medscape Activity. https://www.medscape.org/viewarticle/934463?src=mkmcmr_driv_cust_mscpedu_cv_200729-934463-us-path
2.