
Bone Marrow
Bone marrow basics
Bone Marrow Failure Syndromes
- Aplastic anemia
- Paroxysmal Nocturnal Hemoglobinuria (PNH)
- Pure Red Cell Aplasia
- Agranulocytosis
- Amegakaryocytic Thrombocytopenic Purpura
Bone Marrow in Systemic Disease
- Hematogone hyperplasia
- Promyelocyte hyperplasia
Bone marrow basics
"Liquid" organ without evaluable gross anatomy [1]
- dynamic relationship with lymphoid organs and PB
- different methods of morphologic evaluation
-- trephine biopsy and aspirate smears
- diagnosis often requires ancillary studies
BM evaluation requires cytology, histology, IHC, Flow cytometry, genetics (cytogenetics, FISH,, molecular) clinical information
BM function: site of production of hematopoietic cells
-- Myeloid cells: RBCs, platelets, neuts, eos, basos, monos, dendritic cells, mast cells
--- Lymphoid cells: T-, B- and NK
BM structure
- hematopoietic marrow
-- semi-solid organ in intertrabecular spaces of axial skeletal bones
-- non-hematopoietic marrow: fat,
- potential hematopoietic organs: spleen > liver ? LNs > soft tissue and other sites
Components of hematopoietic BM
- hematopoietic elements: stem cells, maturing myeloid, erythroid, megakaryocytic, and lymphoid elements
- stromal cells: fat cells, fibroblast-like, dendritic, macrophage
- vascular sinuses
Components of complete marrow evaluation:
- BMBx
- BM aspirate (clot prepared from BM aspirate)
- PBS
- Clinical info
- Ancillary studies
Clinical info is essential
- age and gender
- CBC and differential
- medication hx
- previous diagnoses
- lymphadenopathy and splenomegaly
- serology
- infections, esp HIV and hepatitis
- anemia-related studies (iron status, B12, folate)
BM aspirate
- aspirated liquid containing tiny fragments of fractured trabecular bone and fat particles (spicules)
- used to make air-dried smears
- Wright-Giemsa, iron stain, FISH and NGS
Dry tap scenario
- no or poor aspirate obtained either due to BM fibrosis, packed marrow, or poor technique
- touch imprint preps of fresh marrow core (WG-stain and FISH)
- BM core can be pureed or teased apart with needle to release single cells (cytogenetics or flow cytometry)
How to approach aspirate smears
- review and count in excellent areas of smears
- obtain count from several different areas/slides
- routine cases: 200 cell counts
- 500 cell counts in Any MDS, borderline AML, borderline Myeloma
Bone marrow biopsy technical aspects
- should be taken perpendicular from cortical bone surface
- formalin-based fixation: heavy metals improve nuclear features (B5, B+, Bouins AZF)
- decalcification:
- strong acids
- buffered acids
- EDTA
- paraffin embedding
- thin sections (2 um)
Marrow composition
- cellular elements (~50% of space)
-- hematopoietic cells at carious stages of maturation admixed with storomal cells
- adipocytes (~50% of space)
Cellularity (%) = (Area with cellular elements) / (Areas of cellular elements + adipocytes)
- avoid subcortical fatty marrow, crushed or hemorrhagic areas, vascular spaces, empty spaces (air bubbles)
Thought experiment [1]: Marrow cellularity and differential diagnosis
- Hypercellular + high blood counts
--- leukemic hematologic neoplasm
--- appropriate marrow reaction (eg infection)
Hypocellular + low blood counts
--- BM failure (eg aplastic anemia)
--- exogenous marrow suppression
Hypercellular + low blood counts
--- ineffective hematopoiesis
--- PB cell destruction
Hypocellular + normal counts
--- sampling artifact
Medium-power (20x) architecture: marrow microarchitecture
- erythroid and myeloid form clusters
- M:E ratio = 2:1 - 4:1
- immature myeloid elements adjacent to bone trabeculae, granulocytes away from bone
- megakaryocytes do not cluster
40x evaluation: detailed morphology
- meg number and morphology (normally 2-4 per 40x field)
- myeloid maturation (nuclear segmentation, abundant pink cytoplasm)
- erythroid maturation
Bone marrow stroma
Fibroblasts/reticular cells
Mesenchymal stromal cells (MSCs) - a part of the hematopoietic stem cell niche
-Support self-renewing proliferation of CD34 positive hematopoetic progenitors - hematopoietic stem cell "niche" component
Bone marrow collagens Reticulin fibrosis – type 3 (branching) collagen, reticulin stain,
- Internal control: perivascular Collagen fibrosis
- type 1 (nonbranching) collagen, trichrome stain
- internal positive control: osteoid seam
sites of hematopoiesis [1]


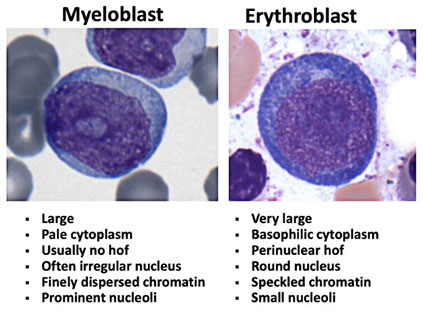



Normal trabecular bone

Type 1 collagen fibrosis

Normal aspirate counts

BM cellularity and age


Megs should normally have 2-4 nuclear lobes, although ok to have a few strange ones

IHC BM [2]

Bone Marrow Failure Syndromes
Aplastic Anemia
1-2 new cases / year / 1 million people (rare)
No gender or race differences in the USA
Marked geographic variability
>1 case in a family rare
Presentation related to pancytopenia:
- bleeding (thrombocytopenia)
- fatigue, other nonspecific symptoms (anemia)
- infection uncommon at presentation
- bimodal age distribution, one peak in the 20s, and one in the 60s (usually due to MDS)
Divided into:
1. Idiopathic aplastic anemia
- accounts for 70-80% of AA
- decreased numbers of stem cells
- impaired responsiveness to growth factors
- in most cases, damage is immune-mediated
2. Secondary aplastic anemia
- Radiation, Drugs and chemicals (cytotoxic agents, benzene), idiosyncratic reactions (chloramphenicol, NSAIDS, antiepileptics, gold, others), Viruses (EBV, hepatitis [non A through G], Parvovirus [rare], HIV [rare]), immuno diseases (eosinophilic fasciitis, hypogammaglobulinemia, thymic neoplasia), pregnancy
Micro:
- PB findings: pancytopenia with normal RBC morphology, macrocytosis may be present, reticulocyte index is low
- BM findings: hypocellular bone marrow, dyspoiesis, if any, is only erythroid (dysplastic neutrophils or megakaryocytes strongly suggests hypocellular MDS rather than AA)
Clonal abnormalities in AA
- clonal cytogenetic abnormalities in 12% of cases
- usually small, may be transient
- monosomy 7 has higher risk of morphologic MDS or AML
Using NGS panels, mutations indicating clonal hematopoiesis reported in 70-85% of AA
- in adults, approaches 100% with WES (whole exome sequencing) or WGS (whole genome seq)
Grading [3]:
Severe AA
- marrow cellularity < 30%
- At least two of:
-- ANC <500/mm3
-- Platelets <20,000/mm3
-- Reticulocytes <1% (<40,000/mm3)
-- No other hematologic disease
Moderate AA
- Pancytopenia
- Do not fulfill severe criteria
DDx:
- inherited pancytopenia syndromes (FA, DKC, esp in younger pts)
- hypoplastic MDS / AML (beware of dysplastic changes, carefully correlated with cytogenetics and FISH) - blast count is critical!!!
- lymphoma (consider doing IHC or flow (esp HCL)
Tx:
Bone marrow Transplantation
- immunosuppression - ATG, CsA, steroids, other
- growth factors
Px: natural history: moderate disease may recover spontaneously, severe disease fatal in days to months
- recent 5-year survival: 75% with immnuosuppresion or immnuotherapy
Proportion of successfully treated pts will relapse
Long-term AA survivors at risk for PNH, MDS, and AML
Aplastic anemia. Some eosinophilic stroma in background, very little hematopoiesis [3]

Mechanism of aplastic anemia. For some reason, some HSPCs are resistant to T-cell attack

MCC of impaired bone marrow production

Paroxysmal Nocturnal Hemoglobinuria (PNH)
First described in 1866, William Gull, in a patient with dark urine noted only in the morning
Acquired clonal hematopoietic disease
- characterized by a defect in GPI glycolipid anchor
- required for the localization of many proteins to the plasma membrane
Median age: 40 years, M:F 1:1, 1 in 100,000
Patients present with intravascular hemolytic anemia (anemia with normal RBC morphology, reticulocytes may be increased, decreased haptoglobin, hemoglobinuria)
- thrombosis (20% of cases in the US)
- Hepatic vein (Budd-Chiari, cerebral vein, abdominal vein)
- Bone marrow failure
PNH and AA
1/3 of PNH pts will present previously with diagnosis of AA
1/3 of PNH patients will eventually develop AA
1/3 remain hemolytic
Pathogenesis
Somatic mutations in PIG-A gene at Xp22.1
- enzyme required for synthesis of GPI moiety
- >100 mutations identified
- leads to absence of GPI anchored proteins
Normal: very small % of PIG-A mutated cells
PNH: selective expansion of PIG-A mutated clone
- Lack of CD55 and CD59 leads to lysis of RBCs and activation of platelets
- PNH type cells also show decreased colony formation in vitro
GPI- Anchored Proteins
Complement: DAF (CD55), MIRL (CD59), C8-binding protein
Immune-related: CD16 (FcgR IIIa), CD58 (LFA-3), CD14, CDw52 (Campth)
Enzymes: acetylcholinesterase, Leukocyte Alk Phos.
Receptors: Urokinase receptor, Folate receptor
Other: CD48, CD66
Labs: (was initially diagnosed only on clinical grounds)
- 1930s: Hemolysis, thrombosis or pancytopenia with positive Ham's test (enhanced lysis of RBCs in acidified serum)
- 1990's: Hemolysis,thrombosis, or pancytopenia with "PNH-type" cells detected by flow cytometry
Flow analysis: Looks for loss of GPI anchored proteins
- Guidelines: use >1 antibody
- examine both RBC and WBC (WBCs may be less prone to spontaneous lysis, which is helpful to monitor the proportion of abnormal cells)
- can use to monitor patient therapy
Micro:
BM: cellularity variable from markedly decreased (AA-PNH) to hypercellular
- Dyspoietic changes may be present
-- be careful!! Hypocellular MDS should be considered
Cytogenetics:
Typically normal, but abnormal cases described (possibly MDS?)
Tx:
Complex - varies c clinical presentation
Immunosuppression - May improve cytopenias – thrombosis management (much of the mortality is due to fatal clots)
-- New! Eculizumab (Solaris) is effective anti-C5 complement inhibitor (very expensive) - WORLDS MOST EXPENSIVE DRUG!!!
- Bone marrow transplant may be considered, usually only in severe cases
Px:
Median survival 10 to 15 years - With flow cytometry now diagnosed much earlier - Low risk of transformation to acute leukemia (less than 3%)
PNH - GPI anchors [3]

PNH Flow - gated on granulocytes, CD24 is GPI-anchored protein, FLAER is Fluorescent Labeled Aerolysin, a fungal toxin that binds the GPI anchors, lower right shows small population of cells negative for FLAER and CD24 [3]

Pure Red Cell Aplasia
Rare, a few hundred cases reported More common in women Median age of onset 60 years
Defined as
(1) anemia
(2) reticulocytopenia
(3) absent erythroid precursors
-Neutrophils, platelets typically normal -Typically little to no dyspoiesis -Normal cytogenetics
__________________________________________
Differential diagnosis Self-limited -Transient aplastic crisis (parvovirus B19) - Transient erythroblastopenia of childhood
Hereditary red cell aaplasia
- Diamond-Blackfan syndrome
Associations Thymoma (classic association! Pure red cell aplasia goes away if excise thymoma)
Neoplasms (CLL, others)
Autoimmune disease
Viral (Pavo B19, hepatitis, HTLV, EBV)
Pregnancy
Drugs (antiepileptics, azathioprine, chloramphenicol, sulfonamides, isoniazid, procainamide, others) Idiosyncratic
Tx
Supportive (transfusions)
Immunosuppression (ATG, CsA, etc)
Treat underlying disorder (Thymoma, CLL, etc)
- >80% respond to therapy or resolve spontaenously
- low risk of transformation to acute leukemia
Agranulocytosis
Peripheral blood: absence of neutrophils and precursors, usually no monocytes, no or mild anemia, no thrombocytopenia
Bone marrow: absence of granulocytic precursors
Many drugs implicated, direct toxicity, immune mediated (like haptens)
Antineutrophil antibodies often present
- nonspecific, also present in vasculitis, autoimmune disease, chronic autoimmune neutropenia
Cytotoxic T cells response may also be present
May truly be autoimmune in absence of prior drug --> "Pure White Cell aplasia"
Px: Untreated, mortality greater than 90% Therapy: withdrawal of drug, growth factor therapy
Amegakaryocytic thrombocytopenic Purpura
Thrombocytopenia secondary to absence of megakaryocytes in the marrow
Very rare end case reports, few small series - Drug-related or autoimmune - Treated with immunosuppression
Fanconi anemia 1927 - Falconi reports three brothers with fatal aplastic anemia and congenital abnormalities
- pancytopenia, hyperpigmentation, skeletal defects, small stature, hypogonadism
Hematologic findings Essentially all patients will develop hematologic abnormalities - At birth, but may be normal, or show only macrocytosis - Pancytopenia develops usually between 5 to 10 years (median age 7) - May be present in adults -as late as 56 years!
Lab tests: Defined by sensitivity of chromosomes to certain damaging agents Diagnosis established by: diepoxybutane (DEB), mitomycin C (MMC)
Micro: Hypocellular marrow, resembling AA - May show display dyspoietic changes - Small, often transient clonal side of genetic abnormalities recorded - May present with MDS or AML
Genetics: Very heterogeneous - 21 genes identified to date - Almost always autosomal dominant - rare XLR cases (2%)
- BRCA2 is one of the genes implicated in Fanconi anemia (aka FANCD1)
PX: Increased cancer risk, at risk for development of MDS, AML (52% by 40 years) - Risk greater than 5000 times over general population - Increased risk for a solid malignancies (squamous cell carcinoma, hepatic tumors)
Median cancer free survival: 29 years - Bone marrow transplant is curative for hematologic abnormalities, but increased cancer risk remains - Growth factor, androgen therapy
Table of defects in Fanconi anemia, 1/3 with no abnormalities!!! [3]

Fanconi anemia. Chromosomal abnormalities caused by dieproxy butane (DEB) or Mitomycin c (MMC) [3]

Genes in Fanconi anemia [3]

Mechanism of Fanconi anemia

Bone Marrow in Systemic Disease
Hematogone hyperplasia
Seen post chemotherapy, post stem cell transplant, immune thrombocytopenic Purpera, Copper deficiency, viral infections, metastatic malignancy
Flow immunophenotype: Dim variable city 45 Very low to low side scatter CD10+, CD19 +
- Partial dim CD20 - Partial CD34 and TdT
Hematogone hyperplasia - blue circles: hematogones; pink circles - mast cells [4]
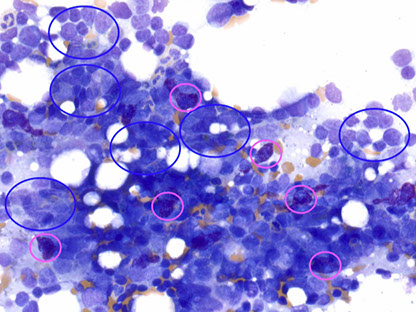
Hematogones in different stages of maturation

Hematogone stages table [4]


Hematogone hyperplasia flow


Promyelocyte hyperplasia
Response to exogenous and indigenous GCSF
Danger of misdiagnosis of AML
Promyelocyte Bulge
- Sepsis or posty G-CSF
- Versus APL: usually some maturation,lack of folded / bilobed nuclei
--- peripheral blood: toxic granulation,Dohle bodies
Promyelocyte hyperplasia [4]

Megakaryocyte Hyperplasia
In setting of increased megakaryocyte destruction or turnover (ITP or post chemotherapy)
- May show mild clustering
Meg hyperplasia [4]


Plasma cell hyperplasia
Reactive plasmacytosis can significantly exceed 10% Morphology can be mildly atypical (binucleation,enlargement) - Etiologies: infection,autoimmune,other
Plasma cell hyperplasia [4]

MonoMAC Syndrome
Rare autosomal dominant syndrome associated with monocytopenia, B and NK cell lymphopenia, mycobacterial (Mycobacterium avium complex MAC), viral (HPV warts), and bacterial opportunistic infections, and the virus infection-induced cancers
- pulmonary alveolar proteinosis
- Progression to MDS/AML
Micro: PB: anemia, monocytopenia, mild large granular lymphocytosis
- BM: patchy hypocellularity, trilineage hematopoiesis, mild dysgranulopoiesis, monocytopenia, focal caseating granuloma
Genetics: inactivating mutations in one of the two parental GATA2 genes is responsible
-12 distinct mutations in GATA2 gene have been identified
- GATA to deficiency syndrome autosomal dominant, variably penetrate MonoMAC syndrome, Emberger syndrome, familial AML/MDS, dendritic cells, monocyte, B (+ hematogone), and NK cell deficiency
Treatment: bone marrow transplant's are currently the only treatment
Causes of Non-malignant dyspoiesis
Avoid MDS diagnosis if potential secondary cause
-- Beware of making the diagnosis in young patients!!!!!!!! Vitamin/micro nutrient deficiencies: copper (ring sideroblasts, vacuoles), Zinc excess (ring sideroblasts), vit B12/folate dificiency Infections: HIV (multilineage dyspoiesis)
Toxins/drugs: EtOH (acanthocytosis, ring sideroblasts), MMF (pelgeroid neutrophils), recent (<6 mo) chemotherapy or radiation therapy (INH--> ring sideroblasts), arsenic/heavy metal toxin
Autoimmune/rheumatologic Congenital: dyserythropoiesis due to increased RBC fragility, baseline dyspoiesis and bone marrow failure syndromes, X-linked sideroblastic anemia, Pearson marrow-pancreas syndrome)
Neoplasms: involving marrow (esp myeloma, LGL, HCL)
post-chemo dyspoiesis

HIV-AIDS-related dyspoiesis [4]
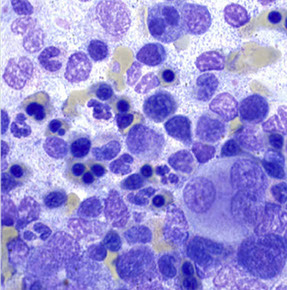
Post chemotherapy bone marrow Week one: complete application - Mixed hematopoiesis absent
- proteinaceous fluid, not fat
- fibrinoid necrosis
- prominence of residual stromal cells, histiocytes, lymphocytes, plasma cells, mast cells
Week2+:early regeneration
- islands of erythroids, mild dyserythropoiesis
- left-shifted myeloids
- megakaryocytes clustered, hypolobated - increased hematogones
- mild reticulin fibrosis
Week 1 Post chemo [4]

Week 2 post-chemo

Coagulative marrow necrosis
-fever, bone pain, cytopenias, leukoerythroblastic reaction -marrow plus/minus trabecular bone -May fibrose with resolution Aspirate: granular basophilic background, loss of cell detail Core: eosinophilic granular material, loss of cellular detail
Differential diagnosis of coagulative marrow necrosis Neoplastic: ALL most common, other leukemia's, lymphomas, carcinoma, neuroblastoma Non--neoplastic: sickle cell anemia (acute chest syndrome), infection, vascular obstruction (DIC, infarct), anti-phospholipid syndrome, acute GVHD following BMT
Coagulative necrosis [4]

Copagulative necrosis

Serous Atrophy
"Gelatinous transformation"
- response to severe malnutrition/cachexia
- hypocellular marrow with loss of hematopoietic cells
- homogenous eosinophilic material surrounds, replaces fat cells


serous atrophy H&E
Amyloidosis
Esprit: pink to purple masses of homogenous material Core biopsy: waxy homogenous eosinophilic material
Amyloid

Hemophagocytosis
May be seen an infection Nucleated-cell hemophagocytosis is more specific for HLH than red cell hemophagocytosis
Hemophagocytosis in HIV infx [4]

Granulomas
Histiocyte inclusions: non-specific findings that can be seen in storage disorders - Gaucher disease: wrinkled silk inclusions, similar cells seen in setting of increased turnover such as CML - Niemann Pick: lipid inclusions, seen in other storage disorders, hyper lipidemia; do not confuse with lipid granulomas
Poorly formed granulomas and seen in immunosuppressed host, fungi, atypical mycobacterium, malignancy, storage disorders
well formed necrotizing granuloma - infx (esp TB, histoplasma)

lipogranuloma

Niemann-Pick inclusions

Granuloma-aspirate, Sarcoid or drug (amiodarone)

well formed granuloma

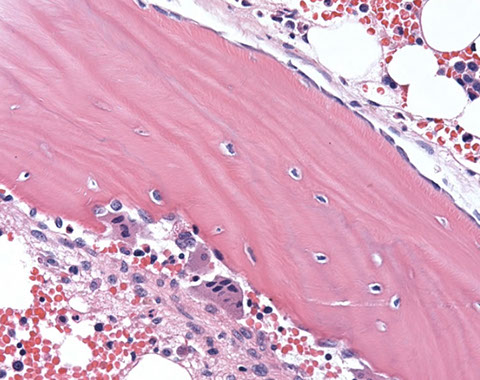
Osteitis fibrosa cystica due to ESRD/dialysis-related secondary hyperparathyroidism
Micro: complex trabecular architecture of variable width -replacement of hematopoietic space by fibrosis -deep invaginations of trabeculae lined with osteoclasts -visible unmineralized osteoid seam
DDX:
- radiologic – metastatic disease, plasma cell myeloma – clinical: primary hyperparathyroidism, renal failure/secondary hyper PTH Histologic: Paget's disease of bone, primary myelofibrosis with osteosclerosis
osteitis fibrosa cystica, circled area and arrow with increased osteoclastic activity [4]


Cystinosis leading to ESRF and renal osteodystrophy
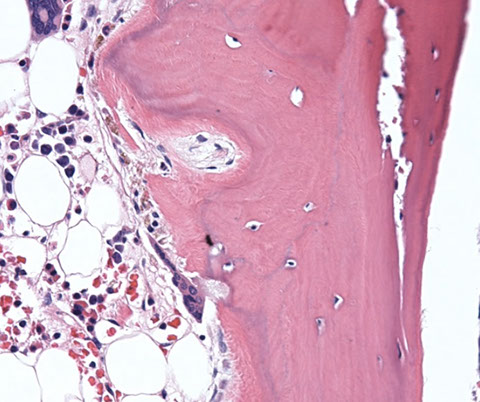
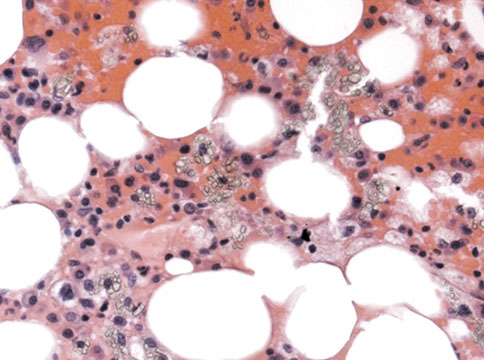


Increased osteoclastic activity
Histiocytes filled with crystals
Histiocytes filled with rhomboid cysteine crystals, which were birefringent with polarized light, which caused renal failure and osteodystrophy in this patient
Increased osteoclastic activity on one side of the trabecula with osteoblastic rimming on the other side
3 - 4
<
>
References
1. Hasserjian R. Introduction to Bone Marrow Interpretation. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=417647306
2. Weinberg O. AML https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=417689596
3. Cook J. Bone marrow failure syndromes. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=417377173
4. Gratzinger D. Bone Marrow Manifestations of Systemic Disease. https://www.society-for-hematopathology.org/web/education-virtual-curriculum-view.php?video_id=417210349
5.