
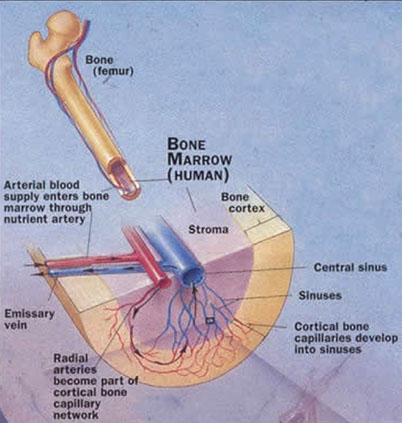
Bone Embryology and Anatomy
Bone Pathology
Bone cysts
- simple bone cyst
- aneurysmal bone cyst (ABC)
Eosinophilic granuloma (Langerhan's histiocytosis)
Osteomyelitis
Achondroplasia
Osteoporosis
Osteopetrosis
Osteomalacia
Hyperparathyroidism
- Brown tumor
- Osteitis fibrosa cystica
Osteitis deformans (Paget's disease)
(Polyostic) Fibrous dysplasia
Myositis ossificans - see Soft Tissue
Benign Tumors
Osteoma
Osteoid osteoma
Osteoblastoma
Giant cell tumor (Osteoclastoma)
Giant cell (Reparative) granuloma - see Salivary Glands, Oropharynx, Teeth
Peripheral Ossifying Fibroma - see Salivary Glands, Oropharynx, Teeth
Osteochondroma (exostosis)
Enchondroma
Chondroblastoma
Chondromyxoid fibroma
Periosteal chondroma
Osteopoikilosis
Aneurismal bone cyst
Ameloblastoma - see Salivary Glands, Oropharynx, Teeth
Osseous dysplasia - see Salivary Glands, Oropharynx, Teeth
Malignant Tumors
Osteosarcoma (osteogenic sarcoma)
- Extraskeletal osteosarcoma (ESOS)
Ewing's sarcoma / PNET
CIC-DUX4 sarcoma
BCOR-CCNB3 sarcomas
Chordoma
Chondrosarcoma
Bizarre Parosteal Osteochondromatous proliferation (BPOP; Nora's lesion)
Adamantinoma
Malignant fibrous histiocytoma
Joint Pathology
Osteoarthritis
Rheumatoid arthritis
Seronegative spondyloarthropathies
Psoriatic arthritis
Ankylosing spondylitis
Enteritis associated arthritis (Inflammatory bowel diesease
Reactive arthritis
Infectious arthritis
Adult Still's disease
Gout
Pseudo-gout
Synovial chondromatosis (osteochondromatosis)
Pigmented Villonodular Synovitis (PVNS)
Bone Embryology and Anatomy
Bones can be flat, cuboidal or tubular
- tubular bones have 3 anatomic regions: epiphysis, metaphysis and diaphysis
-- epiphiphysis is the fat end of the bone
-- metaphysis is where the bone narrows; the physis (growth plate) makes up the majority of metaphysis in dude/ttes still growing
-- diaphysis is the bone shaft (between metaphysis at both ends)
"Apophyses" are sites of growth in long bones (trochanters of femur) and at at sites of tendon attachment
Most bones have a cortex (with periostium and endostium, made of cortical [compact] bone) and a medulla (with trabecular [cancellous] bone)
- the cortical and trabecular bone can be arranged either as woven (unoriented fibers) or as lamellar (sheets) bone
-- woven bone is made rapidly and is seen c most pathology (reactive/neoplastic)
-- lamellar bone is stronger and comprises almost all of a mature skeleton; it is less cellular than woven bone and is made slowly
Bone matrix made of 1/3 osteoid, 2/3 minerals, and is considered type I collagen
- hydroxyapatite makes it hard
- major Ca2+ store
Normal bone structure and physiology
Bone is formed by osteoblasts, which synthesize the structural matrix proteins of bone (collagen, osteonectins, osteocalcin) and alkaline phosphatase, which appears to cleave pyrophosphates and allow for mineralization. Bone is removed by osteoclasts,
multinucleated cells derived from blood monocytes, which synthesize acid phosphatase (involved in dissolving mineral). Osteoclastic activity releases a number of collagen fragments, including the amino acid hydroxyproline and crosslinked peptides (pyridinium crosslinks, N-linked telopeptides). Although bone is often considered a static structure, about 20% of bone matrix and mineral turns over each year. Bone status is determined by the balance between formation and turnover
Cells:
OsteoBlasts - Build matrix with unmineralized type I collagen, which attaches to other bony/cartilagenous structures with a blue cement line
- osteocytes are osteoblasts that got stuck in their own matrix; connect to one another through canaliculi; regulate minerals and mechanotransduction
OsteoClasts - bone Crushers (clast - Gr to break (rocks into littler chunks)
- are MNGCs that get signals to clast bone within their resorption pits
*** Blasts Build, Clasts Crush ***
OsteoClast activity occurs before OsteoBlast activity
(You eat before you work)
Endochondral Ossification
- cartilage mediated
- chondroclasts in first trimester clear out the cartilage mold (anlagen) made by precursor cells of the mesenchyme, making a medullary canal
- at the same time, osteoblasts in the midshaft (diaphysis) deposit cortex beneath periosteum, creating a primary center of ossification
- at the epiphysis there is a secondary center of ossification where endochondral ossification is occuring
- a layer or plate of anlage is trapped bwt the 2 centers of ossification, forming the growth plate or physis
- chondrocytes in the growth plate eventually atrophy and the physis is vascularized to bring nutrients to osteoblasts making osteoid
Involved in:
1) Embryologic long bone formation
2) Linear growth @ epiphyseal plates
3) Fracture Callus
- fracture callus has hemorrhage and marrow necrosis after a couple days; after 2 weeks there is prominent revascularization and cellular proliferation which can look neoplastic if bx'd
Steps:
First, the chondrocytes form a cartilage model
Mineralization lag: time between the deposition of the osteoid (nonmineralized bone matrix) and mineralization, which usually takes ~10 days
Then osteoclasts and blasts replace cartilage c woven bone
- woven bone can be:
1) Immature (not stress oriented, seen in fetal development);
- osteocytes appear very close together, and there are no nice lamellae on polarized microscopy, and enlarged osteocytes
2) Pathologic (fractures, tumors, Paget's dz, etc)
-- is the result of haphazard intertwining of collagen matrix
Woven bone gets subsequently remodeled to lamellar (mature) bone:
1) Compact (cortical) - stress oriented, ie femoral shaft
- layering of collagen around Haversian (vascular) canals; irregularities can be seen as dark blue lines on H&E, also known as "cement lines"
- Osteon (Haversian system): Haversian canals for blood supply, concentric lamellae
2) Trabecular (cancellous) - more elastic than cortical, eg distal femoral metaphysis
Zones of endochondral ossification:
1) Reserve zone
2) Proliferation zone
3) Hypertrophy zone
4) Mineralization zone
5) Primary Spongiosa
Intramembranous Ossification
- Non-cartilage mediated (no anlagen)
- type of ossification seen in skull and other flat bones (scapula, clavicle, sternum, vertebra, jaw, pelvis)
Type of bone seen in flat bones and distraction osteogenesis
Osteoblasts from a fibrous layer directly build this type of bone w/o anlagen
Periosteum
Attached to the cortical bone surface by collagen fibers, known as the fibers of Sharpey
2 layers:
1) Inner Cambium layer
- responsible for new bone formation and increaseing the width of the bone in kiddos
- in adults, this layer gets activated by infection, trauma and neoplasms causing a periosteal reaction or even new bone formation
2)
Remodeling
3 control mechs:
1) RANK (Receptor Activator for NF-Kb) - on osteoclast precursors
2) RANK-L - on osteoblasts and marrow stromal cells
3) OPG (osteoprotegerin) - receptor on osteoblasts [and other cells] that binds RANK-L so it can't activate osteoclasts
- -formed by inc [cellular] of b-catenin from WNT proteins binding LRP5/6
Whether bone is made or clasted depends on RANK-L / WNT ratio
NF-Kb activated when RANK-L binds RANK
Calcium and Bone Metabolism
Calcium physiology
> 99% Ca2+ in body in bones; about 1% of this is regulated by osteocytes and is freely exchangeable with serum calcium. Of non-skeletal calcium, most is in extracellular fluid (concentration about 5 mmol/L), with very little intracellular calcium (< 1 mmol/L).
- In extracellular fluid, calcium exists in 3 forms. Free (ionized) calcium makes up about ~1/2 of total calcium; it is physiologically active and hormonally regulated within a narrow range. Protein-bound calcium (mostly to albumin) makes up ~2/5 of total calcium, but is physiologically inactive. The amount of protein-bound calcium varies directly with protein levels and is also affected by pH; acidosis displaces calcium from protein, increasing free calcium, while alkalosis has the opposite effect. Complexes of calcium with small anions comprise the remaining 1/10 of total calcium; also inactive.
Hormonal regulation of serum calcium
In normal individuals, PTH and calcitriol (1,25 dihydroxy vitamin D) work together to maintain free calcium concentration at normal levels. Calcitriol is the active form of “vitamin” D; after unregulated addition of a 25- hydroxyl group in the liver, renal
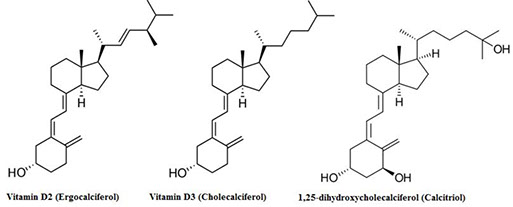
1-hydroxylation is regulated by PTH and/or low phosphate. The net effect of PTH is to increase serum free calcium and lower phosphate, while calcitriol increases both calcium and phosphate concentrations. Calcitonin, produced by parafollicular (C) cells in thyroid, lowers serum calcium by increasing renal excretion of calcium and phosphate and inhibiting bone reabsorption. In humans, states of either overproduction or underproduction of this hormone are not associated with abnormal free calcium concentration in blood, nor with obvious bone disease, suggesting that calcitonin has little physiologic role. PTH-related Peptide (PTHrP) is a 141 amino acid protein; of the 13 N-terminal amino acids of PTH and PTHrP, eight are identical. PTHrP has many of the same biological effects as PTH. PTHrP is produced by most cells, but in greatest amounts by fetal parathyroid, squamous epithelial cells and breast epithelial cells. It is involved in bone formation in the fetus, calcium mobilization in the mother during pregnancy and lactation and transplacental transport of calcium; its physiologic role in other states is less certain. Vitamin D metabolites have become important tests over the last few years, as the high frequency of vitamin D deficiency and its importance have been recognized. Vitamin D2 (ergocalciferol, from diet) and vitamin D3 (cholecalciferol), produced from action of sun on 7- dehydrocholesterol, are first converted to 25-hydroxy forms by the liver; levels of 25-OH D (sum of both forms) reflects total vitamin D precursor available, and is widely used to detect vitamin D deficiency. 25-OH D is activated in the kidney by 1-a-hydroxylase (regulated by PTH and low phosphate) to produce 1,25 (OH)2 D, which is usually not low in vitamin D deficiency. The boards sometimes show vitamin D structures (a steroid ring with the middle 6-carbon ring opened); 25-OH D actually has 2 hydroxyl groups, while 1,25 (OH)2 D has 3 (derivatives of cholesterol, which has 1).
"The D is for Dehydroxylated (-OH groups) ? "
Tests of calcium and bone metabolism
1. Calcium and phosphate – total calcium MC test; interferences include chelating agents (such as EDTA from a lavender top tube) and gadolinium (in MRI contrast), both of which cause falsely low results. Total calcium is directly related to albumin concentration, as well as measuring changes in free calcium. Free calcium is measured by ion selective electrodes; specimens must be handled to prevent changes in pH which will alter free calcium concentration.
2. PTH – produced as 84 amino acid peptide, then cleaved in circulation to several fragments. The most abundant includes amino acids 35-84 and is felt to be inactive. A longer fragment (7-84) represents a minority of circulating “PTH”, especially in patients with renal failure; it binds to different PTH receptors that have the opposite effect of PTH. Most assays for “intact” hormone measure both intact PTH and this 7- 84 fragment, although newer assays for “biointact” PTH measure only the intact hormone. At present, it is not clear which of these two types of assays (or another approach measuring the two forms separately) is preferable. PTHrP does not cross-react in PTH assays. PTH must be interpreted in light of serum calcium; high calcium should inhibit PTH production, while low calcium should cause increased levels.
Disorders of calcium metabolism
1. Hypercalcemia is usually due to primary hyperparathyroidism or malignancy, which together are responsible for about 99% of cases with increased free calcium. Hemoconcentration can increase total calcium.
- Causes of hypercalcemia: Hemoconcentration, malignancy (paraneoplastic syndrome or secondary to bone destruction by tumor, e.g. prostate), Primary hyperparathyroidism, Milk-alkali syndrome, Drug effect, Other endocrine abnormalities (thyroid disease, acromegaly)
a. Primary hyperparathyroidism is inappropriate overproduction of PTH, usually by a parathyroid adenoma. (80%), parathyroid ca (5%), primary parathyroid hyperplasia (15%), possible MEN1/II assoc
- usually mildly symptomatic or asymptomatic; nephrolithiasis (5%) and overt bone disease (< 1%) are the major significant clinical findings. Increases in calcium are stable over long periods, usually between 10.5-12 mg/dL, serum inorganic phosphate low normal / mildly decreased, alk phos inc, inc PT, normal renal function.
b. Malignancies produce hypercalcemia in two main ways. More commonly, tumors produce PTHrP, causing high calcium and low phosphate are the result. This most commonly occurs with squamous carcinomas (lung, head and neck, esophagus) and with breast and renal cell carcinomas. Tumors metastatic to bone can also cause hypercalcemia by locally activating osteoclasts; myeloma is a typical example. Hypercalcemia in malignancy is more severe (50% >13 mg/dL), rises relatively rapidly
and is more commonly symptomatic (confusion or coma, polydipsia, polyuria). PTH is typically suppressed below the lower limit of normal.
c. Rare causes include ectopic vitamin D production in sarcoidosis or other granulomatous diseases and in some B-cell lymphomas, which causes high calcium and high phosphate; immobilization; and lithium carbonate.
2. Hypocalcemia is about as frequent as hypercalcemia. In many cases, total calcium is low, but free calcium is normal, due to decreased albumin; in many series, this causes 80-90% of “hypocalcemia.” Decreases in free calcium are most commonly seen in chronic renal failure (lack of calcitriol), vitamin D deficiency (more commonly associated with hypophosphatemia and/or bone disease than hypocalcemia) and malabsorption; all of these can cause secondary hyperparathyroidism, with much higher PTH (often over 10 x normal) and more severe bone disease than in primary hyperparathyroidism. Magnesium deficiency, due to renal losses (diuretics, alcohol, cisplatin, amphotericin) or diarrhea, causes hypocalcemia and hypophosphatemia due to inhibition of PTH release and actions. Hypoparathyroidism (low calcium and high phosphate) is usually due to surgical removal of the parathyroid glands, autoimmune destruction, congenital lack of parathyroids and thymus (Di George syndrome), or parathyroid infiltration (hemochromatosis).
Radiology of Bone Lesions
Lytic on X-Ray
Type I: Geographic (can put your hand through)
A: Well-defined, sclerosis
DDx: Bone cyst, Brodie abscess, Cartilage lesions (Chondroblastoma, Chondromyxoid
fibroma, Enchondroma), Fibroxanthoma, nonossifying fibroma*, Fibrous dysplasia
B: Well-defined, no sclerosis
DDx: Giant cell tumor*, Bone cyst, Cartilage lesions (Chondroblastoma, Chondromyxoid fibroma, Enchondroma), Fibrous dysplasia, Myeloma/metastasis
C: Ill-defined
DDx: Chondrosarcoma, Enchondroma (active), MFH/Fibrosarcoma, Giant cell tumor, Osteosarcoma, Metastasis/Myelom
Type II: Moth-eaten (can put fingers through) or Type III: Permeative (can't put fingers through) - are mostly malignant
DDx: Ewing sarcoma, Round cell tumors, Malignant fibrous histiocytoma/Fibrosarcoma, Osteomyelitis*, Osteosarcoma, Langerhans cell histiocytosis (LCH)*, Metabolic (Hyperparathyroidism, Severe Osteoporosis)*, Metastasis/Myeloma
POLYOSTOTIC LESIONS, BENIGN
• LANGERHANS CELL HISTIOCYTOSIS (LCH)
• ENCHONDROMATOSIS
• FIBROUS DYSPLASIA
• HEREDITARY MULTIPLE EXOSTOSES (HME)
• OSTEOMYELITIS
• PAGET DISEASE
• NEUROFIBROMATOSIS (TYPE 1)
• ANGIOMATOUS LESIONS
POLYOSTOTIC LESIONS, MALIGNANT
• METASTASIS
• MULTIPLE MYELOMA
• HEMANGIOENDOTHELIOMA
EPIPHYSEAL/APOPHYSEAL LESIONS
DDx: Chondroblastoma, GCT, , Subchondral cyst/intraosseous ganglion, Infection, Langerhans cell histiocytosis, ,Osteoid osteoma/osteoblastoma, Clear cell chondrosarcoma






Bone Remodeling



Fracture callus c endochrondral ossification


Woven Bone


Bone Pathology
Bone cysts
Simple bone cyst
- aka unicameral bone cyst (UBC)
MC in first 2 decades; can present as pathologic fx or be asymptomatic
- in kids in upper femur or prox humerus
Imaging: usually in metaphysis, abutting epiphyseal plate
- may see characteristic "fallen fragment" of bone floating in a cyst
Micro: cyst lined by thin layer or fibrous CT; congealed fibrin can calcify and form cementum; sometimes see GC's
DDx: aneurysmal bone cyst
Tx: aspiration of fluid and inject corticosteroids
UBC


ABC
1-crop-u41993.jpg?crc=156240058)
ABC
Fluid-fluid level ABC

ABC
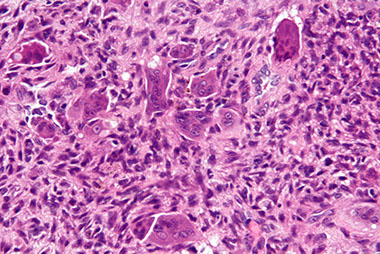
Aneurysmal bone cyst (ABC)
- usually an impressive, "benign" expansile lesion that destroys bone; etiology unknown, prolly a rxn to interosseous hemorrhage
- seen in pts <20 yo, presenting c pain, swelling, MC in metaphysis
-- can compress nerve root if goes vertebral and grow very fast
Imaging: MC in metaphysis of long bones or posterior aspect of vertebra, "soap bubble" characteristic
- CT shows thin rim of sclerosis at margins, and can have fluid-fluid levels (double meniscus 2/2 old blood c lighter overlying watery fluid, or can have soap-bublle appearance
Micro: if intact, see cavernous blood-filled spaces lined by fibrous septae
- septae made of loose / edematous tissue c spindle cells, giant cells, capillaries and thin strands of osteoid or bone + mixed inflam
- can have lots o mits, but not that much atypia
**
Dont confuse reactive, metaplastic part c a bone forming lesion (osteosarcoma or fibrous dysplasia)
- don't miss an underlying lesion (not impossible)
- don't confuse c giant cell tumor of bone
**
Genes: 70% of primary (but not secondary) lesions have USP6 gene chromosome on 17p13.2 (meaning they may actually be unusual lesions (nodular fasciitis has same MYH9-USP6 17;22)
Tx: Exision vs curettage
- no need to irradiate
Px: Rare recurrences
Eosinophilic Granuloma
- aka Langerhan's histiocytosis
- see also Dermatopathology Cancers
Lytic lesion of bone almost exclusively in kids, which can be multicentric, usually presenting c pain
- can be in any bone: skull, ribs, long bones
Imaging: variable, lytic, well-defined lesions
- can have a hole-in-hole appearance on skull x-ray
- may look malignant, c ill-defined infiltrative appearance
Micro: looks like an abscess at low power, but main thing is (of course) proliferation of Langerhan's cells c folded / groovy nuclei in loose clumps
- has variable number of eos, lymphos, plasma cells, and neuts
- may have eo abscess
IHC: (+) S100, CD1a confirms
DDx: infx or non-specific inflam, though in these, Langerhan's cells generally single and dispersed (not in pools or aggregates)
- Erdheim Chester dz, Rosai-Dorfman, osteomyelitis
Tx: solitary bone lesions can be treated c very low-dose radiation, intralesional steroid injection or surgical curettage
- multifocal lesions may need systemic chemo
Px: generally good
- px worse if involves multiple organ systems
Langerhan's histiocytosis- coffee bean nuclei + eos

Osteomyelitis
95% of "uncomplicated" osteomyelitis caused by Staphylococcus; Salmonella assoc c sickle cell anemia
- MC in metaphysis
- may occur at any age, but kids MC affected, esp those c predisposing condition (sickle cell anemia, DM, IVDA, HIV, immunosuppressed)
- affects long bones in kids, vertebrae in adults
- can be acute, subacute or chronic
Sequestrum - piece of devitalized medullary bone that gets separated from surrounding bone in middle of inflammatory process
Involucrum: rim of reactive, sclerotic bone
Imaging: can have aggressive radiologic appearance, that looks permeating and destroys the medulla, and a periosteal reaction
Micro: granulation tissue c mixed inflam c lots of neuts in the medulla that replaces the medulla
- almost 100% neuts if abscess formation present
- plasma cells seen in chronic reaction
- variable fibrosis
Sclerosing osteomyelitis of Garre - aka periostitis ossificans; is seen in jaw bone with lots of new bone formation obscuring underlying bone structures
Brodie's abscess - small intraosseous abscess where only a limited portion of bone affected which can look malignant on imaging
- Tx: excision
Tx: Abx, surgical draining if established infx
- remove sequestrum if chronic (acts as nidus for perpetual infx)
Complication: SCC (rarely) as a result of chronically draining sinus assoc c long-term osteomyelitis

Osteomyelitis, from PathologyOutlines.com, Dr Wick, showing inflam c osteonecrosis and surrounding sclerosis
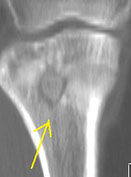
Brodie abscess
Chronic Recurrent Multiple Osteomyelitis (CRMO
Organism not identified
- chronic, recurrent, moves around the patient in different sites of different ages
-- pts seem to get bett, then they get another infx
Micro: Chronic osteomyelitis (c plasma cells), bone remodeling
Achondroplasia
Short limbs 2/2 failure of long bone growth, also bulging forehead, short nose root
- also: trident hand, thoracolumbar gibbus
-- have normal IQ and sperm counts, but shorter life-span (?perhaps due to cardiovascular causes??)
Membranous (ie flat bone [non-cartilagenous]) ossification not affected --> have characteristic large head compared to body
Caused by gain-of-function mutation of FGFR3 (fibroblast growth factor receptor 3), which inhibits chondrocyte prolif, thus, exaggerated inhibition suppresses growth
Usually caused by sporadic mutations in old daddies, but can be AD (double-dominant is lethal in-utero)


Thanatophoric dysplasia
MC lethal form of dwarfism, 1/20k births
- FGFR3 gain-of-function mutation (like achondroplasia)
Short limbs, frontal bossing, macrocephaly, small chest cavity, bell-shaped abdomen
- develop respiratory insufficiency from underdeveloped thoracic cavity, leading to death soon after birth

Dysostoses
Aplasia - absence of bone / digit
Supernumerary digit - extra bone / digit
Syndactyly, craniosynostosis - fusion of bones / digits
2/2 genetically altered transcription factors (Homeobox genes, cytokines, cytokine receptors)
DDx: Amniotic band syndrome
Osteogenesis imperfecta
aka “brittle bone disease”
AD, type I collagen defect, 1/10k affected, failure of osteoblast (high bone turnover and thin trabeculae (ie osteoporosis), may be able to test for collagen type I
Multiple fractures, blue sclera, hearing loss, poor dentition
Fundamental abnormality is too little bone, causing extreme skeletal fragility
- Type I has normal life span, fractures decrease after puberty
Type II is fatal in utero
Genes: Defect in synthesis of collagen type I (COL1A1 and COL1A2) by osteoblast, fibroblast, and other cell types

Osteoporosis
Defined as bone mass density <2.5x SD that of a normal 25 yo individual seen on a DEXA (Dual Energy X-ray Absorption) scan
Decreased trabecular (spongy) bone despite normal mineralization
- osteopenia is 1-2.5 SD on DEXA scan
Despite marked decrease in mineral content in bone, serum calcium, phosphate and PTH are normal
· Bc there no increase in osteoblastic activity, serum alkaline phosphatase normal
2 types
Type 1: Postmenopausal
- inc in osteoclastic bone resorption caused by dec Estrogen which sensitizes the osteoclasts to PTH
- assoc c inc urinary Ca2+ and tooth loss
- see Dowager's hump (vertebral crush fractures) with back pain, height loss and kyphosis (bc Anterior spine is more affected)
- 1% mortality per annum due to femoral neck fx
Type II: Senile osteoporosis
- due to dec Ca2+ absorption, affects both men and women older than 70 yo
- risk factors: age, smoker status, steroid use, white race, thin, lack of weight-bearing exercise, hypogonadism, low calcium intake, prolonged heparin use
- slow bone loss due to inc osteoclastic activity and dec osteoblastic activity
Tx: Bisphosphonates
- estrogen replacement is controversial
Osteoporosis

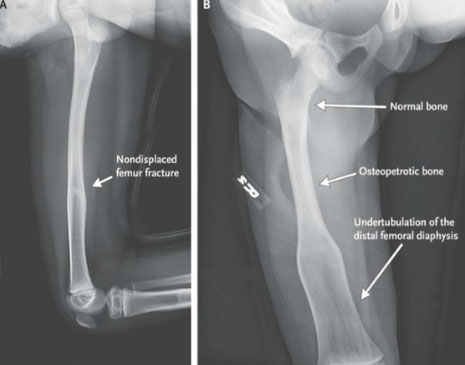
Osteopetrosis
- aka "Marble Bone Disease", Albers-Schonberg dz
- Greek: stone bone
- two forms: AR form that presents in infancy, aka "malignant" osteopetrosis, which is a devastating dz w near 100% mortality by age 10
- adult benign with fractures, cranial nerve compression causing hearing or sight loss or HSmegaly from EM-hematopoiesis (though can be asymptomatic), with normal life expectancy
Osteoclasts fail in resorption of normal bone, leading to weak but thick bones
- have normal Ca2+, PO4-, and Alk Phos (ALP)
- due to Carbonic anhydrase II deficiency
-- without CAII, osteoclasts dont have acid to break down (and thus remodel) bone, causing a build-up of weak bone
Bc marrow is filled with necrotic, calcified cartilage, the osteoclasts lack normal ruffles, and there is no mature trabeculae, pts will be pancytopenic and thus more prone to infx
- patients will have vision and hearing loss bc thick bones in the skull impinge on their respective nerves
Imaging: "Erlenmeyer flask" on x-ray
Pts have anemia and HSmegaly 2/2 reduced bone marrow space and have inc risk of acute leukemia
Genes: CLCN7 (75% of adult subtype)
- all mutations cause osteoclast dysfunction, causing them to be unable to resorb bone, leading to unregulated overgrowth
Tx: vit D, EPO, IFNa, BM transplant (leads to normalization of bone production)
Osteopetsosis: Erlenmeyer flask deformity
Osteopetrosis

Osteomalacia
- aka Rickets (in kids)
Rickets uniquely has: 1) Genus varus, 2) Rachitic rosary, 3) Harrison's sulci, 4) Craniotabes, 5) Growth retardation
- may be an oncogenic (tumor assoc) osteomalacia
Tx: vit D; must use active 1,25(OH)2 vit D if 1a-hydroxylation in kidney is impaired
Qualitative (vs quantitative in osteoporosis) defect in osteoid matrix calcification causing soft (malacia) bones due to vit D deficiency
- main function of vit D is to reabsorb Ca and PO4 from gut in equal proportions to provide adequate bone mineralization
-- thus, loss of vit D leads to dec Ca, raising the levels of PTH (to get more Ca out of the bone) and finally decreasing the serum phosphate (Phosphate Trashing Hormone)
Hyperparathyroidism
PTH has numerous roles in bone / mineral metabolism
Inc PTH can cause osteoporosis, brown tumors or osteitis fibrosis cystica
- PTH functions in osteoclast activation through RANKL expression, inc Ca resorption by renal tubules, inc urinary excretion of phosphates, inc synth of active vit D by kidneys
- In developed countries, hyper-PTH can usually be picked up on routine blood tests, and does not get to the advanced stage of brown tumors
Skeletal changes from inc osteoclast activity
May be primary (autonomous secretion from parathyroid gland) or secondary (to renal dz)
Renal dz has insufficient 1,25(OH)2-D synth, affecting Ca2+ absorption in GI
Hyperparathyroidism - Brown tumors. Right image shows zone of bone resorption in center c defect filled c fibroblastic tisuue, and around the periphery is osteoid-producing area that looks like fibrous dysplasia. Image on left is close of area replacing fibroblastic tissue c lots of osteoclast-like GCs, that may lead to misdiagnosis of giant cell tumor (except GC tumors usually lack fibrogenic stroma)


Brown Tumor
Bone loss causes microfractures and hemorrhage causing macrophage influx and reparative fibrous tissue
Brown from vascularity, hemorrhage and hemosiderin deposition
"Brown tumors" of bone are caused by hemorrhage and hyperparathyroidism
- may also be caused by pseudohyperparathyroidism, but is usually due to parathyroid adenoma, hyperplasia, or rarely carcinoma or chronic renal failure
Osteitis fibrosa cystica
- aka von Recklinghausen's disease of bone
Imaging: erosion of tufts of terminal phalanges and subperiosteal cortical resorption, esp on the radial side of the middle phalanges
Micro: Osteoclasts line cystic spaces (tunneling), which also have a very fibrous stroma, inc formation of woven bone, and peritrabecular fibrosis, MNGCs
Labs: Inc Ca and ALP, low PO4
- hyperparathyroidism: dec Ca, inc PO4 if renal etiology and normal PO4 if extrarenal etiology
Osteitis fibrosa cystica


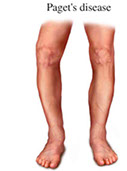
Osteitis deformans
- aka Paget's disease (of bone)
BOTH osteoclastic and osteoblastic activity inc, causing abnormal bone architecture in either one or multiple bones
- MC in late adulthood (eighth decade), lots of pts are asx
- cause uncertain, probably genetic and envtl; may have viral etiology
- anterior bowing of femur and tibia from weight bearing, and may result in chalk stick-type fractures
- skin overlying the affected bone is usually warm 2/2 inc vascularity
3 phases:
1) Hypervascular / osteolytic
- inc in osteoclastic activity with osteoclasts c 20+ nuclei can make bones appear radiolucent (osteoporosis circumscripta)
2) Intermediate: osteoblasts predominate and form weak, "mosaic" bone with loss of corticomedullary demarcation
3) Quiescent phase in which osteoblasts decline and irregular "cement lines" formed
Labs: Normal Ca, PO4, and PTH; very INC ALP
Key terms are inc hat size and hearing loss (nerve impingement similar to osteopetrosis)
- peripheral skeleton is rarely affected
Mosaic pattern bone and long-bone chalk-stick (snapped carrot) fx seen in the sclerotic phase; other phases have less specific morphology
- can acquire high-output heart failure due to inc blood flow from AV shunts
- at slightly higher risk of getting Osteosarcoma; but GC tumors are frequently seen
Tx: Bisphosphonates and Calcitonin

Osteitis deformans


Fibrous dysplasia
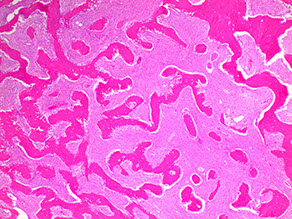
Common. Usually in early adolescence, stops enlarging at time of growth plate closure
- Fibroblasts, collagen, and irreg bony trabeculae replace normal bone (in many bones, but esp femur, ribs and face); unable to make mature bone in multiple places (polyostic) or in a single place (monostotic) which lacks a lining of osteoblasts and is contained w/in a bland fibrocellular stroma and curved bony pattern
- usually in teens / young adults (polyostotic usually in younger pts)
-may be caused by G-protein mutations
- one form of this dz is McCune Albright syndrome (also has cutaneous pigmentation and precocious puberty)
- Mazabraud's syndrome = polyostotic fibrous dysplasia and soft tissue myxomas
Imaging: intramedullary lesion (not cortical) c various degrees of ground-glass opacity (depending on amt of bone there); femoral neck can have classic "shephard's crook" deformity or bowing
- there is little soft-tissue rxn to the lesion
- should still have periosteum above lesion (bc is b9)
Micro: hypocellular fibrocellular stroma c gland oval spindle cells with disoriented trabeculae (Chinese characters / alphabet soup) made of woven bone without a clear border of osteoblasts made directly by the stroma
- commonly see round, calcified cementum-like spherules (esp in the face)
- no cytologic atypia (from video reference below)
- 1/5 have nodules of hyaline cartilage that look like disorganized growth plate
IHC: neg MDM2 (vs LG- osteosarcoma)
DDx: Osteofibrous dysplasia
Genes: somatic gain-of-unction mutation of GNAS1 (also seen in pituitary adenomas) that constitutively activates Gs promoting cellular proliferation
Good case and video:
https://apps.pathology.jhu.edu/bone/case-1/
Fibrous dysplasia


Fibrous dysplasia c ground-glass appearance

Non-ossifying fibroma (NOF)
- looks like benign fibrous histiocytoma, fibrous cortical defect
Micro: osteoclast-like GCs that can be storiforming
- xanthoma cells
Imaging: metaphyseal, geographic IA, eccentric
Bone tumor metastasis
Mets to bone are way MC than primary bone tumors
Tumors that metastasize to bone:
- Prostate (MCC; is blastic)
- Thyroid
- Breast (Both lytic and blastic)
- Lung (is Lytic)
- Kidney (Renal Cell Carcioma)
*** PT Barnum Loves Kids!!***
*** BLT with Kosher Pickles (BLT with KP) ***
Breast Lung Thyroid Kidney Prostate
General bone tumor stuff
- the nuclei often are polarized away from the bone-forming surface and are eccentrically located, and show fine chromatin pattern with prominent nucleoli
Normalization phenomenon: resemblance of tumor cells to normal osteocytes
Benign Bone Tumors
Osteoma
aka Bony Island
See a small (usually 1-2 mm, but can be larger) new piece of bone growing on another piece of bone superficially, which can be protruding or grow into the medulla
- mimics normal bone (made of dense mature lamellar and woven bone)
-- deposited in a cortical-type pattern rimmed c flat osteoblasts and lacking cartilage
- usually in the mandible (skull)
Assoc c Gardner's syndrome (osteomas, colon polyps, lipomas)
Tx: doesn't need tx unless causes sx
Osteoid Osteoma
***bone pain at night relieved by aspirin***
MC in diaphysis of proximal tibia and femur in men <25 yo; although can occur anywhere; may present as a limp
- usually <2 cm (osteoblastoma is larger, but has similar histo)
- although strange, it is a pretty common condition
Imaging: Round lucent nidus (1.5 cm) surrounded by thick, sclerotic bone (target sign)
- CT can help locate the nidus, which can be difficult to find sometimes
Micro: Osteoblasts and osteoclasts surrounded by small, interlacing trabeculae of woven bone and surrounded by vascularized loose CT c dilated capillaries
- neoplastic osteoblasts appear b9
- causes a pronounced reactive formation of bone around lesion
Tx: excision, radiofrequency ablation (must destroy the nidus!!)
- Prostaglandins cause the pain, which is why NSAIDS relieve it...

Osteoid osteoma
Osteoblastoma
NOT self-limited (unlimited growth potential!!!), No nocturnal pain, NOT relieved by aspirin, MUST be >1.5 cm by definition
- big brother to osteoid osteoma
- more rare than osteoid osteoma, also seen in young adults
- MC on vertebrae than appendicular
When seen on appendicular skeleton, usually metaphyseal and diaphyseal; on vertebra are consistently on the posterior elements, not the body; in the jaw forms around the root of a tooth (oral pathologists call it a cementoblastoma)
Imaging: >2 cm c defined margin c or w/o sclerosis
- can show new periosteal bone (1/2) or cortical destruction (1/5) mimicking malignancy
- vertebral lesions c well marginated lucency in vertebral arch
Micro: Osteoblasts surround interlacing trabeculae of woven bone
- must have trabecular for osteoblasts to rim around
- thus, same histologically as osteoid osteoma, BUT is larger and found in the spine (though also seen in prox tibia and femur) and usually goes into the medullary (cancellous) compartment
Several variants: cystic, cartilage-forming, aggressive, and pseudomalignant
- the way to differentiate aggressive osteoblastoma from osteosarcoma is presence of osteoblast rimming and lack of significant atypia and mitosis
Tx: excision c bone grafting
- vs osteoid osteoma, osteoblastoma tends to be progressive (not self-limited)
Osteoblastoma


Giant Cell Tumor
- aka Osteoclastoma
Rare (1 in 1 million); MC in epiphyseal end of long bones (just like chondroblastoma, and the rare clear cell chondrosarcoma), esp around the knee of 20-40 yo
- is a fairly common lesion
- can only be seen in pts c skeletal maturity (***Gotta grow up to be a Giant***)
Although benign, is locally aggressive (10% have malignant transformation) and can met (even if it looks b9!)
- 1-2% mets to lung (which can be surgically excised)
- nuclear features of the MNGCs are identical to the mononuclear cell population in the tumor
- MNGCs are evenly distributed throughout the lesion within small spindle cells (a mix of macrophages and mesenchymal cells)
- NO atypical mits, but regular mits common [?]
Imaging: "Double bubble" or "soap bubble", usually on both epiphysis and proximal metaphysis
- can look malignant on radiology (in 1/4, cortex destroyed and extends into ST), though generally has no tumor matrix production or periosteal production
Micro: Spindle-shaped cells c MNGCs with uniform scattering
- mononuclear cells and GC's have similar nuclei and uniformity of cytologic features, can have 50-100 nuclei per cell, lacking atypia
IHC: H3F3B +
DDx: other lesions c MNGCs = brown tumor of hyperparathyroidism (looks identical, need to check parathyroid / renal status), chondroblastoma, solid aneurysmal bone cyst, giant cell reaction, non-ossifying fibroma, giant cell rich osteosarcoma, brown tumor
Genes: "telomeric associations": telomeres of normal chromosomes joined back to back
- neoplastic osteoclasts have inc levels of RANKL, promoting prolif of osteoclastic precursors into mature osteoclasts
Tx: complete surgical excision
- can give RANKL inhibitor to stabilize if cannot be surgically resected
- may give Gleevec
Px: 1/4 recur, rare mets (3%)
- can turn malignant if recurs, esp if recurring in same site as previous resection that was previously treated with radiation; bx shows frank sarcoma c highly cellular spindle cells (doesn't have the features of avg run of the mill giant cell tumor of bone)

Giant cell tumor


(Giant cell) Reparative Granuloma
Benign, reactive lesion seen in the face or distal extremities (hands / feet) of 10-25 yo that presents as swelling / pain
- unknown cause
Imaging: round / oval lucency c fine trabeculations and well-defined margins
Micro: spindle cell fibroblastic stroma and scattered lymphs and MNGC clusters distributed in the stroma and aggregating around blood and foci of new bone formation (osteoblastic rimming)
- may see cortical thinning, but no cortical destruction or new periosteal bone (?)
DDx: can mimic giant cell tumor of bone, but has: more irregular distribution of GCs, stromal cells more spindled than oval, stroma makes collagen and often osteoid
Tx: curretage and bone grafting
Px: may have high local recurrence that may best be tx'd c amputation if they are destructive

(Giant cell) Reparative Granuloma
Osteochondroma
- aka solitary osteocartilagenous exostosis
MC benign bone tumor; 3M>F
Presents as a hard, painless mass that has been there for years
- usually solitary, but if multiple can be part of multiple hereditary exostosis syndrome
Imaging: pedunculated or sessile
- femur (1/3) > tibia (1/5), humerus (1/5), hand and foot (1/10)
- malig transformation suspicious if thickening of the cartilagenous cap or invasion to bone
Grows @ right angle from growth plate and has mature bone c thin cartilagnous cap
- usually in males < 25 yo; around the knee
-- makes sense that it comes from the metaphysis bc those kids are still growing!
Medullary and cortical component is continuous c underlying native bone
- resembles epiphyseal growth plate
Genes: Multiple hereditary exostosis syndrome assoc c germline loss-of-function mutations in EXT1 or EXT2, encoding an enzyme for synth of heparan sulfate GAGs, which may dec diffusion of Indian hedgehog (Ihh), a regulator of cartilage growth, thus disrupting chondrocyte differentiation
Tx: want to have clear margins so stops growing
Only rarely will it transform to chondrosarcoma (usually in syndromes and with multiple lesions)
- usually stops growing at time of growth plate closure

Osteochondroma

Enchondroma
MC form of chondroma (vs juxtacortical chondroma on bone surface); Rare (1% of all) benign bone tumors affecting the hands and feet (65%), and intramedullary metaphysis of long bones (25%) in younger pts (10-30 yo), presenting c pain and swelling
- grossly are usually <3 cm and gray blue and translucent and well-circumscribed nodules of b9 chondrocytes, that can infarct and calcify
Imaging: well-demarcated purely lytic popcorn-like calcifications (stippled radiolucent defects) and ring-calcifications (formed as part of endochondral ossification) oriented parallel to long axis
Micro: pseudolobules of cellular myxoid and cartilagenous tissue broken up by lines of connective tissue filled c vessels, mononuclear cells and MNGCs
- periphery shows dense spindle cell population (stellate cells in myxoid stroma c microcysts)
- 1 chondrocyte per lacuna
- centers of the lobules have immature cartilage c round chondrocytes that may be hyperchromatic and binucleated, but shouldn't have mits
- assess nuclear atypia at 20x by looking for vesicular chromatin, macronucleoli, and irregular nuclear membranes
-- can have some degree of atypia and inc cellularity, but should not have viable lamellar bone of Haversian canal infilatration (dx chondrosarcoma)
Assoc c Olliers (multiple enchondromatosis) and Maffucci's syndrome (spindle cell hemangioendothelioma)
Genes: Heterozygous mutation of IDH1 and IDH2, encoding isocitrate dehydrogenase, producing the oncometabolyte 2-hydroxyglutarate, which diffuses into neighboring cells and causes oncogenic epigenetic changes (transformation by association)
Medullary margins are scalloped and sclerotic (suggesting benign lesion); peripheral margin can be expanded by secondary aneurysmal transformation
Tx: en-bloc excision
Px 20% recurrence rate
Enchondroma of proximal phalynx

Enchondroma of femur


Enchondroma
Chondroblastoma
Rare (1/50 bone tumors) cartilage-forming tumor in the proximal (epiphyseal) humerus and distal femur of young pts (<20 yo; skeleton still immature); 2-3M>1F
Imaging: round lytic lesion c well-defined sclerotic borders
- may see calcifications which show up as fine opacities
- CT/MRI: fluid/fluid levels
- MRI - not typical chondroid characteristics - low/intermediate T2W (95%), extensive surrounding edema, joint effusion (up to 1/2)
Micro: sheets of immature reniform chondroblasts dispersed in an irregularly distributed group of osteoclast-like GCs
- look for foci of chondroid matrix and pericellular network of "chicken-wire" calcifications
- commonly see secondary aneurysmal change (up to 1/3 c ABC component)
- different from GC tumor b/c GCs in GC tumor are more evenly spaced
IHC: S100 (+) (same as giant cell tumor), H3F3A +
Tx: curretage
Px: generally excellent; ~1/5 recur, can have "b9" mets to soft tissue and lung
- mets should raise suspicions on initial dx; should review original dx to make sure it was chondroblastoma
Chondroblastoma - chicken wire calcs


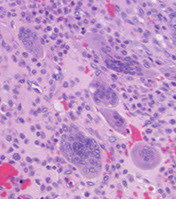
Do not be fooled by the giant cells!!

CMF


CMF

Chondromyxoid fibroma (CMF)
Extremely rare (least common cartilagenous tumor), b9 tumor in young adults (15-25 yo), metaphysis of long tubular bones, presents c dull achy pain
Imaging: Eccentric, often cortical, metaphyseal, high signal on T2 W images MRI, rare matrix
· Usually lower extremity (– 55% around knee, 20%-25% in foot)
Micro: condensation of fibrous change around peripher, myxoid in middle
- can have some stellate atypical-looking cells that appear to be floating
Periosteal chondroma
Mass of lobulated hyaline cartilage in a confined area in the outer cortex made of moderately differentiated cartilage
Osteopoikilosis
AD condition c multiple, bilateral symmetrical enostoses (bone islands)
Enostosis is an incidental lesion that can cause minimal pain found intramedullarly
- if not part of the hereditary dz, an enostosis is an incidental, homogenously dense lesion with spiculated margins that blend c adjacent cancellous bone and are composed almost entirely of lamellar bone (little woven bone)
DDx includes intramedullary osteosarcoma (made of trabecular, not lamellar bone with infiltrative borders and malignant osteoid)
Malignant bone tumors
Osteosarcoma
- aka osteogenic sarcoma
2nd MCC malignant bone tumor (2nd to plasma cell myeloma)
- 20% of primary bone cancers
- classified by site of origin in bone (intramedullary, intracortical, or surface), histologic grade, primary or secondary, and histologic features
Seen in 10-20 yo males around the knee (long bone metaphysis) as painful enlarging mass esp around growth plate at times of growth spurts; but age distribution is bimodal
- older pts c osteosarcoma usually secondary not primary (Paget's dz, rads)
- inc risk c Paget's dz of bone, bone infarcts, radiation, and familial retinoblastoma
-- nearly 40% of osteosarcomas have AD inheritance of retinoblastoma gene
- also p53 mutations
Imaging: Codman's triangle or sunburst pattern (from periosteal elevation) on xray
- this imaging pattern may also be seen c pyogenic osteomyelitis
- MRI can be helpful in determining dz expansion for surgery
Micro: malignant bone stroma with lacy-patterned matrix that at least focally produces osteoid surrounding neoplastic mesenchymal cells and is infiltrating the marrow spaces bwt mature trabeculae
- can see atypical mits (atypical mits NEVER in b9 conditions [?])
- normalization: atpia is less in oseoid islands than in surrounding malignant stroma
Any amount of neoplastic matrix automatically upgrades a lesion to osteosarcoma
MC subtype is conventional high-grade type (made mostly of osteoblastoma; 75-85%); then high-grade secondary osteosarcoma (10%) and surface osteosarcoma (5-10%) - [chondroblastic {makes cartilage}, fibroblastic {no matrix production}, small-call or giant-cell rich]
- LC subtype is intramedullary well-differentiated (1%)
IHC: SATb2 +, MDM2 + (in low-grade osteosarcoma)
- small cell osteosarcoma can be CD99+
DDx: fracture callus, osteoblastoma, tumors c reactive bone formation
Genetics:1p11-13/12, 1q21-22; gene amplification of 8q23 in 50% and 8q24 (MYC) in 40%; can also see overexpression of MDM2, PRIM1, CDK4; and of course 13q14 (RB locus) assoc c retinoblastoma increases the risk 500x
Surface osteosarcomas can be parosteal or periosteal and are MC in women
Parosteal: well-differentiated juxtacortical tumors sticking out like a mushroom on the posterior part of proximal femur; have irregular arrangement of woven (immature) bone lacking osteoblastic lining; and is surrounded by spindle cells
Imaging: no Codman's triangle; center is more dense than periphery, but look out for the posterior mushroom!, invade cortex to medullary cavity
- spindle cells look deceptively like normal fibroblasts (minimal atypical features), they microscopicaaly grow between well formed parallel arranged bony trabeculae
- cartilagenous differentiation seen in ~1/2 cases
- intraosseous part of tumor is completely cut off from the normal bone marrow
- generally has an excellent px, but can get complicated (dedifferentiated) if not completely excised
Periosteal: intermed.-grade with moderately differentiated, atypical (high-grade) cartilage covered in small rim of mesenchymal cells (immature, round to spindle-shaped with central hyperchromatic vesicular nuclei) which is further covered by periosteum, grossly more fusiform
- 2-3% of osteosarcomas, occurring on the bone surface of long bones in young pts
Imaging: fusiform, broad-based radiolucency on bone surface; Codman's triangle
Tx: neoadjuvant tx and en bloc excision
- should grade response to neoadjuvant tx (>95% tumor necrosis is good)
Px: better than that of usual high-grade osteosarcoma; can get mets to lung or other bones
Tx: Limb salvage (as long as tumor doesn't invade neurovascular bundle) with wide excision and adjuvant chemo
Px: Poor
- pathologist asked to estimate the percentage of viable tumor following resection (made by Huvos), wherein >95% tumor necrosis assoc c favorable outcome
Osteosarcoma

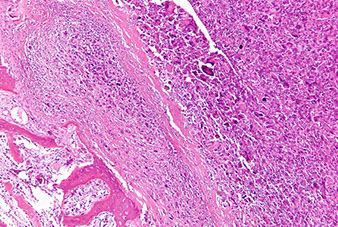



Parosteal osteosarcoma

Periosteal osteosarcoma

Parosteal osteosarcoma
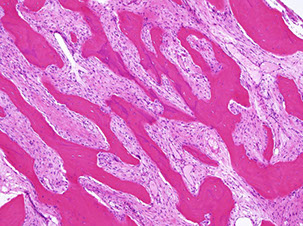
Periosteal osteosarcoma
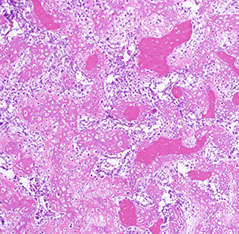
Extraskeletal Osteosarcoma (ESOS)
~4% of osteosarcomas, usually in older pts than regular osteosarcoma, 2M = F, which can arise in almost any ST site (usually the butt and the retroperitoneum)
- may be related to rads exposure
Micro: reticular osteoid matrix c big, pleomorphic spindly to polygonal tumor cells c little bit of red cytoplasm and large dark nuclei
IHC: (+) vimentin (not specific)
- negative desmin, cytokeratin
Tx: Cut it out, rads, chemo
Px: aggressive, high mortality (4/5)
- px may depend on tumor size (>5 cm bad) and mits
Ewing's sarcoma / PNET tumors
Anaplastic, high-grade malignant small round blue cell tumor MC in boys <15 yo
- may present as warmth and swelling in the diaphysis of long bones and in pelvis, scapula and ribs (bone involvement assoc c soft-tissue extension, can occur in any bone)
- another micro feature is Homer-Wright pseudorosettes and spotty necrosis
- 4th MCC bone tumor (after myeloma, osteosarcoma, chondrosarcoma)
- 2nd MC primary bone sarcoma in children
- called Ewing in soft tissue, PNET in bone (?)
Imaging: "onion skin" appearance, saucerization (concave pressure erosion of the cortex when mass stuck bwt periostium and cortex), and Codman's triangle
*** Going out for wings and onion rings***
Micro: uniform small round blue cells, eosinophilic cytoplasm
- monotonous nuclei ,inconspicious nuclei and low mits are deceptively low grade
- occasionally has Rosettes
EM: abundant glycogen
Immunohistochemistry
•Positive: CD99,NKX2.2, FLI1 (~85-90%), ERG (~5%)
•Sometimes positive: neuroendocrine markers, focal desmin, focal keratin
•Negative: TdT, lymphoid markers, melanocyte specific markers, myogenin
Genetics: t(11;22) by rt-PCR or FISH
- EWSR1 gene on 22q12; FLI1 gene on 11q24
- if negative for the mutation, CD99 vairable, and lots of necrosis, may consider CIC-DUX4 sarcoma (see Soft Tissue)
*** Patrick Ewing's basketball jersey was 33 (11+22), and he can FLY! ***
- many other fusion partners have been identified, such as ERF, ETV1, FEV, POU5F1
- the fusion type is of no prognostic significance
Genetics
•EWSR1::FLI1 (85-90%), EWSR1::ERG (5-10%); rare fusions: EWSR1::ETV1, EWSR1::ETV4, EWSR1::FEV
Molecular confirmation should be pursued if possible
•Be careful with FISH (many tumors have EWSR1rearrangements)
•NGS superior
(+) membranous CD99 (gene product of MIC2 gene) - not specific b/c produced by other soft-tissue / small round blue cell tumors
- also has focally prominent cytoplasmic glycogen c PAS/PAS-D
- FLI1 (+ in 85-90%) seen in cases c t(11;22)(q24;q12) translocation
- (+) ERG in cases c t(21;22)(q22'q12)
-- ERG expressed in normal and neoplastic endothelial cells
- NKX2.2 is a homeobox transcription factor that targets EWSR1-FLI1 and reportedly has high sens and spec
DDx: metastatic neuroblastoma (younger pts, neuropil, (+) synaptophysin), embryonal or alveolar rhabdomyosarcoma (usually metastatic [desmin, myogenin]), malignant lymphoma of bone, small cell osteosarcoma, mesenchymal chondrosarcoma
Tx: responsive to chemo after resection and rads
Px: Extremely aggressive c early mets (previously 75% survival at 5-years, now better) = moderate to poor px


Ewing's sarcoma / PNET

A, Ewing sarcoma consists of solid sheets of small, blue, round cells with geographic necrosis and nuclear expression of Friend leukemia integration 1 transcription factor (FLI1) (B).
Desmoplastic Small Round Cell Tumor (DSRCT)
Primarily occurs in children and young adults c predilection for boys
- usually in abdomen, retroperitoneum, or pelvis c widespread serosal implants
- named 2/2 prominent stromal desmoplasia
- has multiphenotypic differentiation, and can be + for markers staining epithelium, muscle, and neural tissue
Micro: tumor necrosis, freq mits, and cystic degeneration common
- glandular and pseudorosette formations can be seen
IHC: express epithelial (EMA and CK), myoid (dot-like desmin positivity), and neual (NSE), WTI (nuclear)
- neg SMA
Genes: EWSR1-WT1 from t(11;22)(p13;q12) and selective WT1 carboxy-terminus reactivity (not dual amino- and carboxy-termini as seen in Wilms tumor)
Px: not good
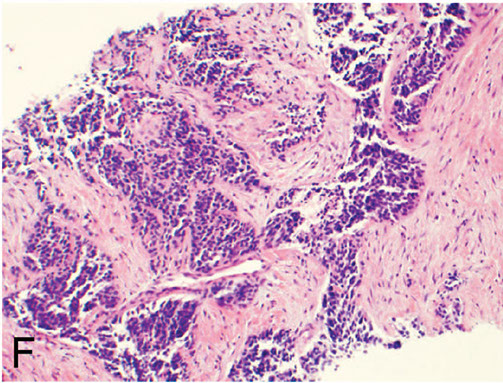
F, Desmoplastic small round cell tumor exhibits nests of round cells separated by a prominent, densely collagenized stroma
CIC-DUX-4 Sarcoma
- type of Ewing-like sarcoma c non-EWS fusion
- for Ewing sarcoma / PNET, or BCOR-CCNB3 sarcomas,
- ST tumor in extremities
Micro: Similar to Ewing sarcoma c nucleoli, may have a little more cytoplasm and lots of necrosis
IHC: + WT1, ETV4
- variable CD99;
Genes: t(4;19)(q35;q13), or t(10;19)
- fusion protein has binding site for TLE protein, just like synovial sarcoma
- upreagulates ETS family of genes
- may suspect in EWS FISH negative Ewing-like tumors
•Novel chromosomal translocation t(4;19)(q35;q13)
•CIC-DUX4dominant oncogene >100-fold enhanced transcription by mutant CIC
•Increased binding of ERM promoter with increased expression of ETS transcription factor gene family (may be translocation partners with EWSR1)
•Transformed NIH 3T3 fibroblasts
Px: Aggressive c early mets


Superficial CIC-rearranged sarcoma
BCOR-CCNB3 sarcoma
Rare (<5%); looks like Ewings, except has lower cellularity and is myxoid
IHC: (+) BCOR
- neg WT1, ETV4
Genes: fusion of BCOR (BCL-6 corepressor) to B3 CCBN3 (an epigenetic regulator), amplifying ability of cyclin B3 to drive cell cycle events
- BCOR loses function and is epigenetically instable
- may be able to do CCNB3 by IHC
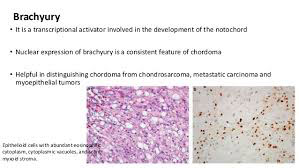
Chordoma
Low- to medium-grade malignancy comprising up to 4% of primary bone tumors that occurs mostly in adults 40-60 yo in the sacrococcygeal region (50%) or skull (30%), can cause constipation bc usually occur in sacrum and can grow large
May originate from notochordal rests at the end of the spine
Micro: Cells c lightly acidophilic, vacuolated cytoplasm in solid sheets or cords or evenly singly floating in an abundant myxoid stroma
- Physaliferous cells: large c pale multivacuolated cytoplasm and prominent nucleoli
- can see moderate atypia, but mits are rare
(+) S100, CKs, and epithelial monocyte antigen; brachyuria (specific)
- negative GFAP
High rate of local recurrence, mets are rare
Px: 65% at 5 years; nobody survives 10 years, has high local recurrence rates, true distant mets occur late in dz and are rare (<10% of pts)
Chordoma



Chondrosarcoma
2nd MCC bone malignancy (25%, excluding MM) in adults; which arise de novo
Malignant cartilaginous tumor MC in 30-60 yo (older) males
- usually in spine, pelvis, scapula, humerus, tibia or femur (more central location [vs peripheral in enchondroma]) that presents c dull pain (benign cartilage tumors don't produce sx usually) over a long period of time
- very rarely (~1%) involve the extremities
Can be primary or secondary to enchondroma or osteochondroma
Primary Chondrosarcoma types:
Intramedullary
· Periosteal/Juxtacortical
· Clear cell
· Mesenchymal
· Myxoid (see Soft Tissue for extraskeletal myxoid chondrosarcoma)
· Extraskeletal
· Dedifferentiated
Secondary Chondrosarcoma types:
Enchondroma
· Osteochondroma
· Paget disease
· Radiation induced
· Miscellaneous
Grossly appears as a glistening mass within a medullary cavity
- can also look like "salt n pepper" or "popcorn-ish"
Imaging: soft tissue mass, cortical thickening, and endosteal scalloping; range from completely opaque to completely lytic
- lesions are malignant if they have >2/3 scalloping in a long bone or invasion to soft tissue
Micro: Islands of cartilage permeating viable lamellar bone; bone made by enchondral ossification
- classified into 3 grades based on cellularity, atypia and pleomorphism
-- most are grades 1 (lobulated; resembles chondroma) or 2 (not lobulated and made of spindle-shaped cells mixed c lacunar cells)
Lobules of hyaline to myxoid cartilage with binucleation, cytologic atypia, increased cellularity
· Grades I (30%): Hyaline cartilage, only mildly atypical
· Grade II (40%): Myxoid change, increased cellularity, atypical
· Grade III (30%): Increased atypia, spindle cell change at periphery of lobules
IHC: (+) S100, D2-40
- negative CK, EMA, GFAP, brachyury
DDx: Chordoma (EMA+, brachyury +)
Genes: t(9;22)
- most have mutations in IDH1 and IDH2
Tx: surgical excision c wide margins
- radiochemo for high-grade tumors
Px: indolent; will recur a number of times before mets
- the higher the grade the more recurrences and higher incidence of mets

Chondrosarcoma grade I
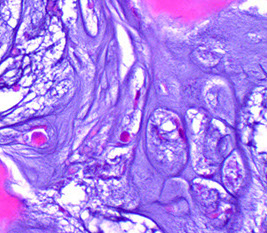

Chondrosarcoma Grade II

Mesenchymal chondrosarcoma
Mesenchymal chondrosarcoma
Highly aggressive and malignant tumor often in the jaws and ribs of young adults
Micro: Biphasic tumor made of well-differentiated hyaline cartilage and a small round blue cell tumor
- small round blue cell component can have areas of spindling, hemorhage, necrosis, and a vascular pattern like a hemangiopericytoma (stag-horn!)
- occurs in different location than normal chondrosarcoma?
- SOX9 - master regulator of chondrogenesis
Gene: HEY1-NCOA2


Mesenchymal chondrosarcoma
Mesenchymal chondrosarcoma (arrows to mesenchymal component
Intramedullary Chondrosarcoma
Symptoms -pain (95% - 99%) and mass (82%); Male > Female (3:2), Avg age 40-45 year; Metaphysis;
· Location – axial/midline: pelvis (30%), femur
(25%), shoulder (15%), ribs/sternum (10%),
vertebrae (7%), scapula (5%)
Genes: IDH mutation
Myxoid chondrosarcoma
(see Soft Tissue for extraskeletal myxoid chondrosarcoma)
Rare variant c monotonous group of small cells c acidophilic cytoplasm and vesicular nuclei arranged in cords or sheets in a myxoid stroma
- M>F
Tx: wide surgical excision
Px: frequently get mets
Extraskeletal myxoid chondrosarcoma

Dedifferentiated Chondrosarcoma
comprise ~1/10 of all chondrosarcoma, abrupt transition from cartilaginous neoplasm to highly malignant spindle cells (look like fibrosarcoma or undifferentiated sarcoma

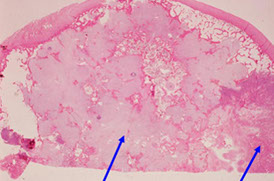
Dedifferentiated chondrosarcoma
Dedifferentiated chondrosarcoma, left arrow is low-grade cartilagenous part, right is dedifferentiated part
Bizarre Parosteal Osteochondromatous Proliferation (BPOP)
- aka Nora's lesion
Rare, aggressive tumor in soft tissue of hands and feet adjacent to bone that resembles osteochondroma on imaging
Arises directly from cortex and lacks the continuity bwt lesional and cortical bone as in osteochondroma
Micro: Lobulated, cellular cartilagenous tissue c calcified fibrocartilagenous tissue at periphery; also c bone and fibrous tissue
DDx: florid reactive periostitis, subungual exostosis
- these are assoc c trauma; BPOP is not, however

BPOP


Adamantinoma
Rare low-grade malignancy of bone in the intercortical part of the anterior tibial diaphysis or sometimes the fibula (50% have both tibial and fibial involvement!)
- has epithelial component that resembles amyloblastoma of jaw (has peripheral palisading and central stellate reticulum); though many features overlap with osteofibrous dysplasia
- usually peaks in teens and 20's, though wide age range
- pt usually presents c chronic leg pain
Imaging: Well-circumscribed, multiloculated intracortical radiolucency (multiple lucencies) c sclerotic margins
Micro: differentiation of multipotent mesenchymal cells causes it to be biphasic with tubular spaces lined by flat or cuboidal cells and compact basaloid nests with a central vascular-looking space
- has interspersed keratin-positive epithelial islands
- can have sarcomatoid transformation c lots of pleomorphism, lots of mits
IHC: vimentin, keratin and Endothelial monocyte antigen stain the epithelial cells
-(+) CK 14 and 19
- neg CK 8/18
Tx: excision [???]
Px: slow growing and indolent, but if tx inadequate has 25% mets to LN or lung
- differentiated adamantinoma (osteofibrous dysplasia type) doesn't met
- high rates of local recurrence
Adamantinoma


Malignant fibrous histiocytoma of bone
Rare (<2%) primary bone tumor in pts >50 yo around knee that presents c pain (from bone expansion) and a soft tissue mass
Imaging: Lytic, poorly defined destructive lesion c permeating moth-eaten margins
- bc cortical bone destroyed, can present c pathologic fx
Micro: biphasic c fascicular fibrous part and high-grade pleomorphic part c storiform pattern
- both parts are infiltrative into marrow spaces around thick trabeculae c unusual forms
- often in center is necrotic one (tumor may arise from area of infarcted bone)
Tx: aggressive surgery c chemo
Px: mets to lung and LN; poor (15% @ 5 yrs)
Joint Pathology
Osteoarthritis
Mechanical wear and tear of superficial articular cartilage with surface fibrillation (surface appears velvety), and then subchondral cysts, osteophytes (bone spurs, new bone and cartilage formation [from remodeling]), and eburnation (polished, ivory-like subchondral bone) once the articular cartilage is completely worn down
--sclerosis (has mature, lamellar architecture), NO ankylosis, Heberden's nodes (DIP), Bouchard's nodes
*** Morning stiffness in Osteroarthritis lasts O-30 min ***
Imaging: osteophytes, sclerotic bone, joint space narrowing, subchondral bone cysts on xray
~ osteophytes: reactive bone formation at joint margin irritates synovial lining causing pain
~~ eventually bone rubs on bone, causing dense sclerosis
Age, obesity, and joint deformity are predisposing factors
- Secondary causes of OA: obesity, ochronosis (alkaptonuria), hemochromatosis, Legg-Calve-Perth (aseptic necrosis of femoral head ossification center), osteochondritis dessicans (trauma to articular epiphysis --> ischemia --> aseptic necrosis and failure of articular epiphysis)
Classic presentation: pain in weight-bearing joints after use, improving w rest, morning stiffness, crepitus
- cartilage loss in knees begins on medial aspect ("bow-legged")
- non-inflammatory condition
- No systemic symptoms
Secondary causes: obesity, ochronosis (alkaptonuria), hemochromatosis, Legg-Calve-Perth, osteochondritis dessicans
*** Morning stiffness: in Osteoarthritis lasts O-30 minutes, but in Rheumatoid arthritis lasts a Really long time (30+ min) ***
Micro: Vertical clefts / fibrillations in remaining cartilage
Articular surfaces missing from surfaces rubbing together, aka eburnation
Thickened areas of cancellous bone beneath eburnation
Microfracture of exposed articular bone
Previous fracture allows synovial fluid to enter, where it is walled off as a subchondral cyst
Tx: acetaminophen on regular basis, NSAIDS, steroid/ hyaluronic acid injection in joints, opioids (last resort)

Rheumatoid arthritis
Morning stiffness that lasts >30 min and improves c use
*** in RA morning stiffness lasts a Really long time***
- symmetric synovial joint involvement and systemic symptoms
-- synovial fluid studies show: dec viscosity, dec C3, inc WBCs
Females > males
Type III Hypersensitivity
~80% have positive rheumatoid factor (anti-IgG ab)
-- synovial cells express an antigen that cross-reacts c B-cell receptors stimulating B-cells to become plasma cells and produce RF (an ab that binds the Fc of IgG)
- anti-CCP ab less sensitive but more specific
-- strongly assoc c HLA-DR4
Features:
Pannus is granulomatous tissue rich in inflam cells and fibroblasts in joint space that proliferates and releases cytokines (TNF-a and IL-1) causing a cell-mediated destruction of articular cartilage and eventual ankylosis (a type IV HS)
- Carpal tunnel from flexor-tenosynovitis or median n compression from swelling
-- Rheumatoid synovitis on micro shows villous formation, hyperplasia/hypertrophy of the lining cells, fibrinoid necrosis and chronic inflam in the synovial membrane (tx'd surgically c physiotx)
Atlantoaxial joint subluxation
Subcutaneous rheumatoid nodules
Ulnar deviation (No DIP involvement)
Swan neck deformity
Ischemic strokes
Caplan's and Felty's syndromes
Pleuritis c pleural effusion, carditis, vasculitis, atherosclerosis
Baker's cysts behind the knees
Anemia of chronic dz (dec Fe and TIBC, inc ferritin)
Rice bodies - free-floating fragments of synovium and cartilage in the joint space
Rheumatoid subcutaneous nodules - Occur in ¼ affected individuals, usually in areas subjected to pressure; in elbow, forearm, occiput
- Central necrosis rimmed by palisading histiocytes
- MCC death: coronary artery dz
Tx: NSAIDS, DMARDS, TNF-a inhibitors, leflunomide, rituximab
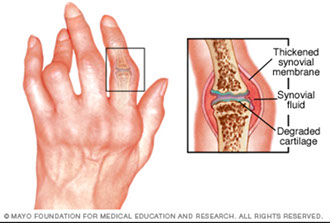

Seronegative spondyloarthropathies
- general term for arthritis without rheumatoid factor (no anti-IgG ab); although has strong assoc c HLA-B27 (which codes for HLA MHC I) and occurs more often in males
- pathologic changes occur in ligamentous attachments and not in synovium; though must involve sacroiliac joints
Includes: Psoriatic arthritis, Enteritis assoc arthritis, Ankylosing spondylitis, Reactive arthritis
*** PEAR ***
Seronegative spondyloarthropathies
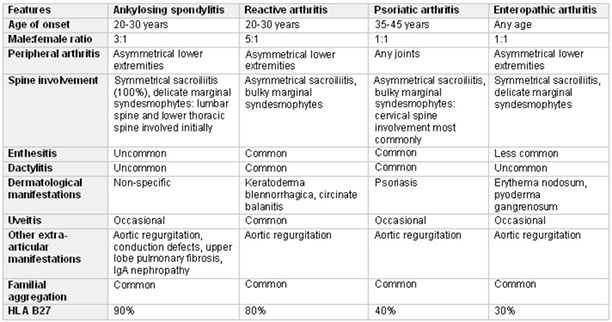
Psoriatic arthritis
Joint pain and stiffness (arthritis) assoc c psoriasis (affects ~30% of pts c psoriasis) which is asymmetric and patchy and inflam of the entheses (ligaments and tendons)
- >80% have a psoriatic nail condition such as nail pitting or loss of nail itself
- usually occurs bwt 4th and 6th decades
- 1/10 pts c psoriasis get psoriatic arthritis
Imaging: Dactylitis ("sausage fingers") and "pencil in a cup" deformities on x-ray, and hands and feet are MC involved
Genes: dz susceptibility related to HLA-B27 and HLA-Cw6 alleles
Tx: NSAIDS (naproxen)
Enteritis assoc arthritis
Arthritis caused by GI infx by Yersinia, Salmonella, Shigella and Campylobacter and others; 2/2 LPS in outer cell membranes that stim immune response
- lasts for about a year and then generally clears
Ankylosing spondylitis
Chronic inflam dz of spine and sacroiliac joints that leads to ankylosis (stiff spine due to joint fusion), uveitis / iridiocyclitis (40% of pts), aortic regurg / aortitis (leading to CHF), problems c cardiac electrosignal conduction
Seen in males in 20-30's; 9/10 have (+) HLA-B27
- worse c inactivity and in morning; better c exercise and hot showers
- Synovitis c enthesitis
- at site of inflam, there is bone destruction / formation c syndesmophyte formation
- bone syndesmophyte eventually bridge gaps bwt vertebrae, forming a rigid bamboo spine
Reactive arthritis
- formerly known as Reiter's syndrome
Asymmetric oligoarthritis 1-4 wks after non-gonococcal urethritis or infx diarrhea, in lower extremities
- classical triad is 1) Conjunctivitis and anterior uveitis, 2) Urethritis, 3) Arthritis (*** Can't see, can't pee, can't climb a tree***)
- may also see oral ulcers, keratoderma blenoohagica (of palms and soles), and circinate balanitis
Bugs that can cause this: Ureaplasma, Campylobacter, Chlamydia, Salmonells, Shigella, Yersinia
*** U CCSSY, can't even see, pee or climb a tree!! ***
Waxes and wanes over 6 months, 1/2 get recurrence
Infectious arthritis
All kinds of bugs can get into the joints during hematogenous dissemination
Septic / Suppurative
MCC is S aureus in older kids and adults; H influenza MC in kids <2 yo; sickle cell pts get Salmonella at any age
- gonococcal arthritis is an STD that presents as a monoarticuar, migratory arthritis c asymmetric pattern, MC in women
*** STD = Synovitis (knee), Tenosynovitis (hand), Dermatitis ***
- viruses can mimic inflam arthritis, but generally resolve quickly
Chronic
TB or lyme dz
- can also be caused by viruses
Juvenile Idiopathic Arthritis (JIA)
Heterogenous groups of dz of unknown cause that occurs b4 age 16 yo and lasts >6 wks
- have systemic dz more commonly than in RA, but no rheumatoid nodules
- ANA is usually (+)
- risk factors are HLA and PTPN22 variants, and damage mediated by Th1 and Th17 cells and by IL-1, IL-17, TNF, INF-gamma
Adult Still's disease
Inflam dz c recurrent high fevers (>39C), rash and arthritis
Rash is maculopapular and non-puritic, affecting the trunk and extremities during febrile episodes




Gouty tophus
Gout
Transient attacks of asymmetric joint pain, swelling and erythema caused by deposition of monosodium urate (MSU) crytals in and around joints
Classic presentation is in the big toe (podagra) after a large meaty meal c red wine
Primary (9/10) or secondary hyperuricemia cause monosodium urate crystals to precipitate
-- other (secondary) predisposing factors are Lesch-Nyhan syndrome, PRPP ecess, dec uric acid secretion, inc cell turnover (leukemia), von Gierke's dz, or dec excretion of uric acid (pts taking thiazide meds)
-- vast majority of primary gout due to inc uric acid biosynthesis from unknown causes
- deposition of crystals in joints stimulates production of cytokines that recruit WBCs
Must aspirate joint to dx
- crystals are needle shaped and negatively birefringent
Acute arthritis shows dense neutrophilic infiltrates that permeate synovium (can see MSU crystals inside neuts)
Chronic tophaceous artritis is 2/2 repetitive acute attacks
- synovium is hyperplastic, fibrotic and thick c inflam cells, forming a pannus that destroys underlying cartilage and bone erosions
- can lead to formation of ankylosis and loss of joint function
Tophi form, which are granulomatous multinucleated giant cells c gouty crystals that occur on external ear, Achilles tendon, olecranon bursa
Tx: aspiration and steroid injection
- chronic gout tx: probenecid for under-excreter
-- Allopurinol for over-producer
--- giving colchicine while on allopurinol may provoke a gouty episode
-- febuxostate is another xanthine oxidase inhibitor which can be used if pt is allergic to allopurinol or has renal dz
Px: 20% of gout pts eventually get
Pseudogout
- aka Calcium pyrophosphate crystal deposition disease (CPPD)
Calcium pyrophosphate crystal deposit in joints, esp the large joints (knee, elbows); chondrocalcinsis (cartilage calcification) of knee
Grossly may see chalky white material in poorly vascularized structures
- blue rhomboid crystals that are weakly positively birefringent (yellow when perpendicular, blue when parallel)
Affects those >50 yo; MC risk factors: hyperparathyroidism and hemochromatosis
- may be hereditary and can be triggered by trauma or surgery
Imaging: Chondrocalcinosis (punctate / linear radiopacities) in articular cartilage and other opacities in periarticular structures and the synovium
Tx: colchicine, NSAIDS
Pseudogout



Ganglion cyst
Small cyst near joint capsule or tendon sheath, usually on joints or wrist, where it is firm, fluctuant and pea-sized
- results from cystic or myxoid degeneration of CT, and thus lacks a cell wall lining
- can be multiloculated, and has joint-like fluid although does not communicate with joint space
- is not related to ganglia of the nervous system, despite name
Micro: Dense fibrous tissue w/o synovial or epithelial lining; inflam can be caused by rupture of cyst


Synovial Cyst
Cuased by herniation of synovium through a joint capsule or 2/2 enlargement of a bursa
- ie Baker cyst in the popliteal space in RA
- synovial lining can be hyperplastic and contain inflam cells and fibrin
Synovial cyst



Synovial chondromatosis
- aka osteochondromatosis
Monarthritic dz c multiple metaplastic hyaline nodules within the articular space found just beneath the synovial lining cells (which were previously sublining cells of the synovium); affecting primarily the larger joints (hip, knee, shoulder)
- large age range; M>F
Pt presents c gradual onset of pain and "locking" of joints, with evidence of possible joint effusion
- the synovial surface is grossly hypertrophied c lots of little nodules
- in the nodules, chondrocytes arrange in clusters along the periphery
-- malignancy assoc c the loss of these clusters and more dispersed chondrocytes and the presence of spindle cells
-- in early dz the nodules are small and entirely cartilagenous
-- in late dz, large nodules occupy enlarged villi and the cartilage undergoes ossification (hence osteochondromatosis) and break free and float around the synovium (loose bodies)
- moderate atypia (binucleation and hyperchromasia) is common and not necessarily indicative of high grade
Synovial osteochondromatosis is when these nodules form by means of endochondral ossification
Imaging: multiple circumscribed opaque calcifications assoc c erosion of articular margins on xray
- CT/MRI shows signal voids w/in membrane (may be necessary if xray normal)
Gene: FN1::ACVR2A found in ~1/2 of cases, confirming the neoplastic nature of this disease
Tx: Synovectomy (in the intrasynovial dz phase); in the inactive phase, joint body excision is sufficient without synovectomy
Synovial chondromatosis


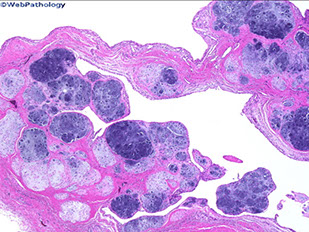

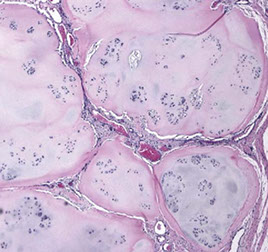
Pigmented VilloNodular Synovitis (PVNS)
Aggressive prolif lesion affecting large joints and causing adjacent bone erosion that commonly affects the tendon sheaths in the finger joints, but also the knee or hips
- the lesion is usually swollen but not really painful and may show signs of internal derangement
-- no systemic sx typically seen
Imaging: peri-jointacular swelling with jointy erosions and lytic defects on xray; intralesional punctate signal voids on CT/MRI
On excision, the joint fluid may be brown and the articular surface can be growing in a fern-like or nodular pattern
Micro: villous hypertrophy of the synovial surface c hyperplastic cells that are filled c hemosiderin
- has a hypervascular membrane c lipidy-macrophages, occasional GCs, and some lymphocytes
Tx: surgical excision
Px: often recurs

PVNS
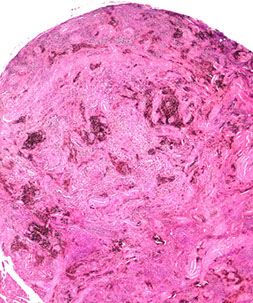

Tenosynovial Giant Cell Tumor
- related to Giant cell tumor of tendon sheath ?? - see Soft Tissue
Group of b9 neoplasms that form in synovial lining of joints, tendon sheath and bursae
- can be diffuse or localized types
Genes: t(1;2)(p13;q37) causing fusion of type VI collagen a-3 promoted upstream of coding sequence of M-CSF gene
- thus tumors overexpress M-CSF which stimulates prolif of macrophages (similar to GC tumor of bone)
Giant cell tumor of tendon sheath, belong to the locaalized variant of tenosynovial GC tumor
