
Adrenal
Anatomy, Embryology and Histology
Adrenal steroids pathway
Cortisol
Congenital Bilateral Adrenal Hyperplasia (CAH)
Hermaphroditism and pseudohermaphroditism
Parasites
Waterhouse-Friderichsen Syndrome
Primary hyperaldosteronism (Conn's syndrome)
Secondary hyperaldosteronism
Adrenal cause of Cushing's syndrome
Ectopic ACTH
Addison's disease
Pheochromocytoma
Adrenal Cortical Adenoma
Adrenocortical carcinoma
Black adenoma
Neuroblastic tumors
- Neuroblastoma
- Ganglioneuroblastoma
- Ganglioneuroma
Paraganglioma - see Nervous System Tumors
Adrenal and paraganglia
Anatomy
Appears pyramidal on the right and crescentic on the left
3 independent blood supply from inferior phrenic artery which forms superior adrenal artery, the aorta gives rise to middle adrenal artery and the renal artery the inferior adrenal artery
- dual blood supply to adrenal medulla from straight fenestrated capillaries (sinusoids) and medullary arteries
- no veins or lymphatics in adrenal cortex, but drains through central vein in adrenal medulla
- the medulla does not extend into the tail normally
Layers of the adrenal cortex and Their Hormones
*** Go Find Rex; Make Good Sex ***
Glomerulosa --> Mineralocorticoids (Aldosterone)
Fasciculata --> Glucocorticoids (Cortisol)
Reticularis --> Sex hormones (Androgens)


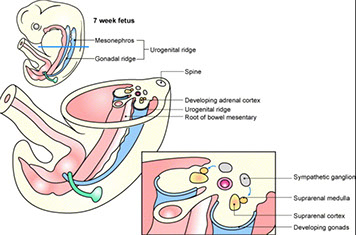

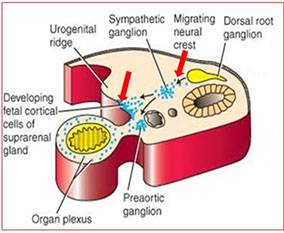
Embryology
5th WGA mesothelium-derived cells go into retroperitoneal mesenchyme at cranial end of mesonephros giving rise to primitive adrenal cortex
- at 7th WGA this mesothelial cell mass invaded by neural-crest derived chromaffinoblast cells that differentiate into chromaffin cells of the adrenal medulla
- thus the medulla is derived from neuroectodermal and the cortex from mesodermal derivation
- at birth the zona glomerulosa and fasiculata develop under control of ACTH produced by fetal pituitary gland
The cortex during fetal life known as fetal cortex, and the big plump eosinophilic cells involute rapidly after birth, leaving behind only the outermost cortex
-



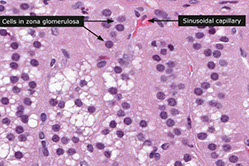



Histology
Zona glomerulosa narrow subcapsular zone c concentrically arranged cells surrounded by stroma c capillaries
- cells have lipid droplets and well-developed sER
Zona fasciculata is the largest zone and has polygonal cells in vertical columns / fascicles perpendicular to capsule, and also have lipid droplets in cytoplasm c cholesterol and its metabolites
Zona reticularis is somewhat thicker than glomerulosa and cells have lots of lipofuscin
The medulla has 2 cell (epinephrine- and norepinephrine- secreting) types surrounded by venous sinusoids
- epinephrine-secreting cells are 4x as numerous as the norepi- ones
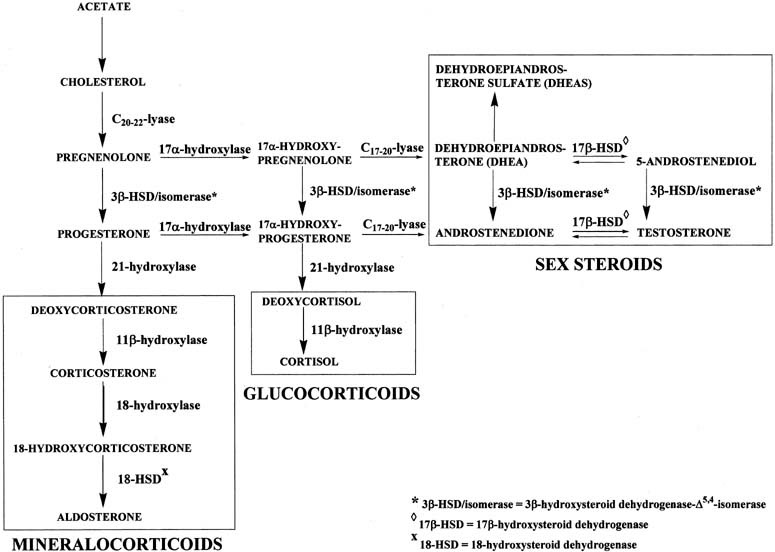
Adrenal Steroids Pathway
From cholesterol to Aldosterone, cortisol, estradiol, DHT, Estrone
Cortisol
Made by zone Fasciculata
***think BBIIG: maintains Bp, dec Bone formation, anti-Inflammatory, dec Immune function, inc Gluconeogenesis/ lipolysis
Regulated by CRH (hypothalamus) which stimulates ACTH release (pituitary)


Adrenal Physiology
Normal Adrenal Function
Glucorticoids, mineralocorticoids and catecholamines are anatomically connected, they are functionally distinct and each has separate regulatory pathways
- states of adrenal hormone excess usually involve only one of the 3 classes of hormones
-- deficiency states may, by destroying the entire gland, affect all 3 hormone types, or (if they are due to problems in the organ producing the factor that regulates adrenal hormone production) only one.
Hypothalamic–pituitary–adrenal glucocorticoid axis
Cortisol is the only endogenous steroid that inhibits CRH and ACTH release
- exogenous steroids (prednisone and dexamethasone) also inhibit CRH and ACTH release
CRH and ACTH released in secretory pulses throughout the day
- highest activity shortly before waking
- Stress also stimulates release of both CRH and ACTH; major factors producing the “stress” response are IL-6 and serotonin
Cortisol
- assays measure total (mostly protein bound) cortisol and may cross-react c some synthetic glucocorticoids (not dexamethasone) and steroid metabolites
- Plasma free cortisol assays not widely used, but are not affected by binding protein levels
- Serum cortisol usually highest at or around rising and lowest at bedtime; measurement of salivary cortisol (proportional to plasma free cortisol) at bedtime is often used to test cortisol overproduction
- normal morning cortisol in an unstressed, ambulatory individual is 5-25 mg/dL, while normal bedtime cortisol is < 3 mg/dL.
-- Cortisol increases during stress; critically ill patients may have cortisol > 200 mg/dL
Urine free cortisol
Very little cortisol excreted to urine before being metabolized, but slight increases in cortisol above the upper reference limit results in marked increases in urine free cortisol
Urine steroid metabolites
- Older assays for 17-hydroxysteroids (measure cortisol, its immediate precursor and their metabolites) and 17-ketosteroids (which measure most androgens, but not testosterone) use chemical tests, rather than immunoassays and are subject to many interferences.
- only situation when useful is if synthetic block is suspected, as
in congenital adrenal hyperplasia or in adrenal carcinoma.
The adrenal medulla is a component of the sympathetic nervous system and its major function is to serve in the synthesis of catecholamines, the most important of which is epinephrine.
- Catecholamine excess may occur transiently with any form of stress, or with drugs that stimulate catecholamine production, such as caffeine
- Prolonged excess of catecholamines usually 2/2 tumor of adrenal medulla
-- In children, neuroblastoma and in adults, pheochromocytoma arise from the adrenal medulla. Pheochromocytoma is a rare cause of hypertension (< 0.1%), often accompanied by tacchycardia, sweating and hyperglycemia.
Urine catecholamines or metabolites (particularly metanephrines) are the MC tests of catecholamine production. Because some pheochromocytomas produce only epinephrine (normally the least abundant catecholamine), even significant elevations may not increase total catecholamine metabolites. For this reason, many recommend measurement of fractionated catecholamines or metanephrines in urine. Plasma catecholamines are not commonly measured because they increase with any type of anxiety (even anticipation of having blood drawn!). Measurement of fractionated metanephrines in plasma has recently been promoted; these assays are probably the most sensitive screening test, but have a much higher rate of false positive results. Because many neuroblastomas do not produce norepinephrine, rather halting the
pathway at dopamine, measurements of HVA and VMA are usually
combined in the initial evaluation and in follow-up of children with this tumor.
Congenital Bilateral Adrenal Hyperplasias (CAH)
- AR; all have enlargement of adrenals due to inc ACTH stim 2/2 dec cortisol; pts get adrenal hyperplasia from inc ACTH
- 17-hydroxyprogesterone elevated in all forms of CAH
Any steroid hormone not requiring the deficient enzyme will be produced in excess.
Labs: document low cortisol and increased levels of the metabolites (based on the clinical features observed)
- Normal cortisol rules out complete deficiency, but low normal may occur with partial deficiencies
- sx of mild androgen excess (hirsutism, menstrual irregularities) often not evident until adrenarche (increased adrenal androgen production around the time of puberty), thus partial deficiency may be asymptomatic until that time (aka “late onset” or “non-classical” congenital adrenal hyperplasia)
-- Measurement of metabolites before and after short term ACTH stimulation can establish presence of a partial deficiency
*** 17, 21, 11 ***
- the first number is for blood pressure, the second number is masculinization, if 1 is hypertenstion (think up arrow) or masculinization (think penis)
1) 17a-hydroxylase deficiency
dec sex hormones, dec cortisol, inc mineralocortioids
- thus pts have HTN, hypokalemia
-- XY have dec DHT --> pseudohermaphroditism (external phenotype is female, no internal reproductive structures [2/2 MIF])
-- XX have external phenotype female c normal internal sex organs, but lack secondary sex characteristics (sexual infantilism)
2) 21-hydroxylase deficiency
MC adrenal deficiency; ~95% of CAH (rest are 11-h def)
- gene encoding 21-hydroxylase is CYP21 [6p21] in the HLA locus
Dec cortisol (inc ACTH), dec mineralocorticoids, inc sex hormones
- masculinization, female pseudohermaphroditism, HYPOtension, hyperkalemia, inc plasma renin activity, volume depletion
Salt wasting can lead to hypovolemic shock in newborns
3) 11B- hyroxylase Deficiency
2nd MCC of CAH
- esp common in North African Jews
Dec cortisol, dec aldosterone and corticosterone, inc sex hormones
Masculinization, HYPERtension (2/2 excess 11-deoxycortisone)
Steroid Hormone Pathways
Hermaphroditism and pseudohermaphroditism
Genotypic sex - determined by the status of the Y chromosome - the presence of a single Y chromosome (regardless of the number of X chromosomes) is definitive of a genotypic male
Gonadal sex - depends on the histologic nature of the gonads. The Y chromosome, in particular the SRY gene, influences the genital primordium to develop into testes; in the absence of a Y chromosome, the gonadal primordium will develop into ovaries
Phenotypic sex - refers to the appearance of the external genitalia, which may be ambiguous
True hermaphrodite

True hermaphrodite
Term implies the presence of both ovarian and testicular tissue, either separately or within the same gonadal structure (ovotestis). This is an extremely rare condition in which the genotype is usually 46, XX but by FISH, transposed SRY gene material can nearly always be found. In some cases, in fact, true hermaphroditism is assoc c 46,XX/46,XT mosaicism. A 46, XY genotype is not assoc c true hermaphroditism
Pseudohermaphroditism
Refers to discordance bwt phenotypic sex and gonadal / genotypic sex. The presumed sex is that indicated by the gonadal histology
- male pseudohermaphrodites have a Y chromosome and testes, but the external genitalia (and often the genital ducts) are either ambiguous or feminized. There are a wide variety of causes, the MC of which are the androgen insensitivity syndromes (testicular feminization).
- Female pseudohermaphrodies have no Y chromosome (usually 46, XX), ovaries and external genitalia that are either ambiguous or male appearing (virilized). Female pseudohermapheroditism is caused by excessive exposure to androgens during fetal development, MC 2/2 CAH
Androgen insensitivity syndrome
- XR, affects genotypic males (46, XY)
- range of severity from testicular feminization at birth to adult infertility, with phenotypes described as complete, partial or mild androgen insensitivity syndrome
- in complete androgen insensitivity, the external genitalia resemble that of a female, and testes typically undescended
- serum testosterone normal to elevated, with normal or elevated serym luteinizing hormone (LH)


Steroid sulfatase deficiency
Prolonged or nonprogressive labor, unresponsive to oxytocin, may be the 1st sign of steroid sulfatase deficiency
- after birth, cholesterol sulfate begins to accumulate in the blood and in tissues
- in skin, it appears to prevent the dissolution of desmosomal attachments (desquamation), leading to the X linked ichthyosis (scoly skin) phenotype
- high incidence of cryptorchidism, with higher incidence of germ cell tumors
- gene for steroid sulfatase (STS) located at Xp22.3
Parasites
MCC in the adrenal is Echinococcus granulosus larva
Waterhouse-Friderichsen syndrome
Acute adrenocortical insufficiency 2/2 adrenal hemorrhage assoc c Neisseria meningitidis septicemia, DIC and endotoxic shock
- acute onset c febrile illness followed by circulatory collapse and death in hours
- adrenal crisis is an acute adrenal insufficiency where cortisol and pressors are needed for support
Waterhouse-Friderichsen

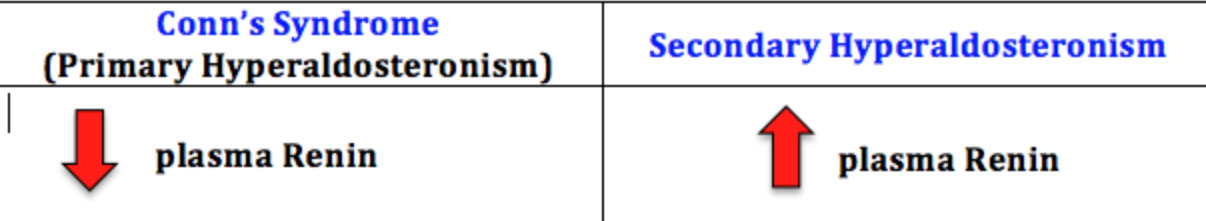
Primary Hyperaldosteronism (Conn's syndrome)
80% caused by solitary adenoma (aldosterone-secreting) which causes HTN, hypokalemia, metabolic alkalosis and low plasma renin
- hypokalemia can cause muscle weakness and paresthesias
- can also see hypocalemia bc alkolosis causes -COOH side groups of albumin to form -COO- which binds calcium and leads to dec free [Ca2+]
- is the cause of as much as 5-10% of hypertension in the U.S., often accompanied by hypokalemia (due to high urinary K losses) and metabolic alkalosis
- High aldosterone and low renin are characteristic
- MCCs are: aldosterone-producing adrenal adenoma, curable by surgery; and essential hyperaldosteronism, associated with increased sensitivity to angiotensin II causing hyperplasia of the zona glomerulosa, which is not cured by surgery
-- In general, aldosterone levels are lower in the latter state (inc sens to angiotensin II) than in former (adrenal adenoma) and aldosterone levels high in only one of the adrenal veins c adenomas
Renin is an enzyme produced by the kidney. An inactive
precursor of renin, termed prorenin, is present in the circulation in high concentrations; conversion of prorenin to renin occurs in whole blood in vitro at refrigerator temperatures. Aldosterone can be measured in either serum or urine; simultaneous measurement of renin and aldosterone in plasma makes it easier to interpret results. The effects of posture and volume must be kept in mind. Renin and
aldosterone levels are highest in the morning and in the upright position; they are lowest in the afternoon and in the supine position. Volume expansion will decrease their levels, while volume depletion will increase them.
Unilateral adenomas can be resected; bilateral should be given spironolactone


Secondary Hyperaldosteronism
Kidney perceives low intravascular volume, leading to overactive RAA system
- may be caused by renal artery stenosis, chronic renal failure, CHF, cirrhosis, or nephrotic syndrome
Labs: high renin and high aldosterone 2/2 renal artery stenosis, accelerated (malignant) hypertension and volume depletion
- If renin concentration higher in the kidney with the narrowed artery (renal vein renin ratio of > 2:1 of affected versus normal kidney), then surgical cure of hypertension is likely.
Assoc c inc plasma renin, inc Na+, inc H2O, dec K+
Tx: correct the underlying cause; B-blocker or diuretic for HTN
Best initial test is plasma Aldosterone: plasma Renin in serum
- most accurate test is to take a sampling of the blood draining the adrenal gland
Ser810Lys Mutation
Missense mutation in the mineralocorticoid receptor that causes early-onset hypertension exacerbated by pregnancy
- usually presents before age 20 yo
Cushing's syndrome
15% caused by adrenals
- adenomas, carcioma, nodular, adrenal hyperplasia
- dec ACTH
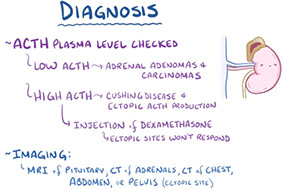
Dx: labs used to both detect Cushing’s syndrome (high urine free cortisol, bedtime salivary cortisol and/or failure of overnight dexamethasone suppression) and helps differentiate 3 causes:
1) pituitary ACTH overproduction [Cushing’s disease]
~60-70% of total cases; suppression with high dose dexamethasone suppression or by localization of increased ACTH production using CRH and dexamethasone to control ACTH production and sampling inferior petrosal sinus blood for ACTH
-- If Cushing’s disease is ruled out, measurements of ACTH can distinguish ectopic production (high ACTH) and adrenal tumors (low ACTH).
2) ectopic ACTH production by nonpituitary tumors [esp. small cell and carcinoid tumors of lung]
3) adrenal neoplasms
ACTH levels are not as useful as TSH, both because episodic ACTH production makes it difficult to interpret and because many adrenal diseases are due to pituitary or hypothalamic dysfunction.
Dexamethasone suppression test (DST) - dexamethasone, a potent glucocorticoid that inhibits ACTH synthesis, is used in differing doses to evaluate the amount needed to inhibit ACTH (and ultimately cortisol) production. While very sensitive for Cushing’s syndrome, a number of factors can cause non-suppression, especially with the overnight test. These include stress, obesity, and drugs that increase dexamethasone metabolism (such as ethanol, phenytoin).
Adrenal stimulation tested by giving 250 mg synthetic ACTH (cortrosyn, cosyntropin). An increase of serum cortisol to > 18-20 mg/dL confirms an intact adrenal. This dose raises ACTH levels to about 1,000 times physiologic, so some recommend much smaller
doses (1 mg).
Pituitary stimulation is sometimes used instead of the above 2 tests. Metyrapone inhibits 11-hydroxylase, causing a fall in cortisol, which increases ACTH and 11-deoxycortisol (by (> 7 mg/dL). Insulin
hypoglycemia should stimulate ACTH and cortisol levels (to > 18-20 mg/dL).
Massive macronodular adrenocortical disease (MMAD)
In adults, nodules >3 mm
Primary pigmented nodular adrenal disease (PPNAD)
Kiddoes, pigmented micronodules < 3mm





Hyperplasia of the adrenal cortex
Usually secondary, primary hyperplasia uncommon
- cortical hyperplasia, can be diffuse or nodular, or combo of both
- the usual cause of hypersecretion of ACTH by a pituitary microadenoma, or ectopic secretion of ACTH by small cell lyng ca (a paraneoplastic syndrome) or other tumors
- causes virilization 2/2 adrenal cortical hyperplasia, entire cortex is brown, and fasciculata is narrow or absent
- clinical syndromes 2.2 hyperplasia of adrenal cortex from excess secretion of glucocorticoids or androgenic steroids
Micro: zona reticularis and fasciculata thickened c reticularis disproportionately increased
Pseudo-hypoaldosteronism
usually 2/2 inactivating mutations in delta-G1226, delta-T1597, C/T1831stop, and delta-A intron-5 splice site
- causes constitutively activated mineralocorticoid receptor, although the mineralocorticoid receptor retains normal activation by aldosterone
Pigmented adrenal cortical hyperplasia
aka primary pigmented nodular adrenocortical dz
- rare cause of ACTH-independent Cushing syndrome
- can be familial, AD and asspc c Carney complex
Gross: variably sized adrenals c multiple brown-black pigmented cortical nodules, which may extend to corticomedullary junction or periadrenal fat
Micro: prominent lipofuscin deposits, myelolipomatous change, lymphocytic infiltrates,
Tx: bilateral adrenalectomy
Ectopic ACTH
15% of all cases of Cushing's syndrome
- nonpituitary tissue makes ACTH
-- ie small cell lung ca, bronchial carcinoids
- inc ACTH



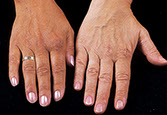

Addison's disease
Chronic adrenal insufficiency or primary adrenal failure 2/2 adrenal atrophy (loss of >90% of the cortex of both adrenals) or dz (TB, metastatic ca, amyloidosis or autoimmune)
Aldosterone deficiency causes hyponatremia, hyperkalemia, metabolic acidosis and volume depletion
Cortisol deficiency causes weight loss, hypotension, hypoglycemia, weakness and decreased resistance to infection; hyponatremia (due to an SIADH-like state) may also be present, but without volume depletion
-- High ACTH may also cause increased skin and mucosal membrane pigmentation.
Primary def of aldosterone and cortisol, which is caused by loss of adrenal cortex
- in developed countries, MCC is autoimmune ("idiopathic")
-- Autoimmune polyendocrine syndrome type I (APS1): chronic mucocutaneous candidiasis, skin abnormalities, dental enamel and nailes, and is assoc c AIRE gene utation (thymus, self-reactive T cells)
-- Autoimmune polyendocrine syndrome type II (APS2): in adults, autoimmune atrophy of adrenals, with Hashimoto's thyroiditis and DM, condition is known as Schmidt syndrome
In secondary adrenal insufficiency, only features of cortisol deficiency are present
Sx: Hypotension, skin pigmentation (a hormonal tanning salon 2/2 Melanocyte Stim Hormone [MSH] biproduct of PrOpioMelanoCortin [POMC] from the pituitary) and vitiligo (from inc ACTH/MSH)
- opposite of hypercortisolism (basically / physiologically[?])
- also see eosinophilia, hypoglycemia, and hyperkalemia
Metyrapone test: metyrapone inhibits 11B-hydroxylase leading to dec cortisol
-- suggested diagnostic approach involves measuring cortisol before (and collecting a sample to save for ACTH measurement) and after stimulation with cortrosyn; cortisol should reach values > 18-20 mg/dL 30 minutes after administration. If cortisol is low, high ACTH confirms primary adrenal insufficiency, while low ACTH is seen in secondary (or tertiary) adrenal insufficiency.
Distinguished from secondary adrenal insufficiency (no pit ACTH production) which doesn't have hyperpigmentation or hyperkalemia
Tx: hydrocortisone, fludrocortisone if pt has postural instability
Pheochromocytoma
MC adrenal tumor in adults
- phaios = dusky, chroma = color
Derived from chromaffin cells (neural crest)
- HTN and/or hyperglycemia may be the presenting sx
***Rule of 90's: 90% benign, 90% unilateral, 90% adults, 90% sporadic, 90% intra-adrenal, 90% have no calcifications ****
Assoc c MEN IIA (Sipple) or IIb, NF1, Surge-Webber, vHL
Most secrete EPI, NE and dopamine which can cause episodic HTN
Gross: usually circumscribed and encapsulated
- cut surfaces typically pale red or red-brown, soft and slghtly bulging
- chromaffin reaction (incubation c potassium dichromate causes tumor to turn brown-black 2/2 oxidation of catecholamines)
Micro: cells large, c abundant cytoplasm, arranged in cords, separated by vascular spaces
IHC: (+) NE markers (CHR, SYN, NSE, CD56, INSM1), S100 (sustentacular cells)
- neg CK
- loss of SDHB seen in ~15% of pheochromocytomas
EM: neurosecretory granules
Dx: inc urinary VMA (from NE), inc plasma catecholamines (metanephrines)
Tx: Phenoxybenzamine (an a-antagonist, nonselective irreversible B-blocker)


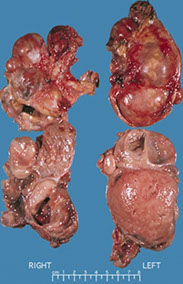
Bilateral pheochromocytoma in MEN2
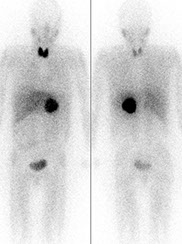
MIBG scintigraphy
Pheochromocytoma with Zellballen pattern



Adrenal Cortical Adenoma
Usually solitary and small, well-encapsulated, homogenous yellow (carcinoma more variegated and have necrosis/hemorrhage)
- palpable lesions are almost always malignant
-- must distinguish benign adenoma from adrenocortical carcinoma and RCC
Common, small and with an orderly layering, like normal cortex, also usually solitary and unilateral
- nonfunctioning adenomas are the MC, and found incidentally
Some syndromes assoc c functioning adrenocortical adenomas:
1) Primary aldosteronism (Conn syndrome) - pts c hyperaldosteronism and have small and yellow tumor, micro shows solid nests of large clear lipid-aden cells and spironolactone bodies (small eosinophilic concentrically laminated bodies related to tx); excision of tumor is curative, adjacent gland does not atrophy
2) Cushing syndrome: hypersecretion of glucocorticoids, causing obesity, hypertension, purple striae, amenorrhea, imptence, osteoporosis, diabetic glucose tolerance curve, mental problems
- uninvolved cortex of both adrenals are atrophic 2/2 reverse feedback
3) Adult adrenal virilism: female that develops hirsutism and receding hairline, baldness, acne, beard, enlarged clitoris deep voice, amenorrhea
- find high urinary 17-KS
- usually has brown cut surface
Gross: circumscribed, encapsulated tumors made of adrenal cortical cells
- should be differentiated from accessory cortical nodules
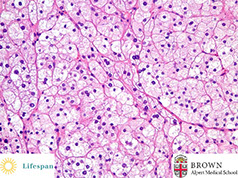
Histo: adrenal cortical adenoma at the right resembles normal adrenal fasciculata. The capsule of this benign neoplasm is at the left. There may be minimal cellular pleomorphism within adenomas
- can be a subtype c myolipoma, a black adenoma, corticomedullary mixed tumor, adrenocortical oncocytoma, or a myxoid adrenocortical neoplasm
Designated as high grade if more than 20 mitoses per 10 mm^2
IHC: Steroidogenic factor 1 (SF-1) the the most reliable marker of cortical origin in evaluating a renal mass (sens 98.6%, spec 100%)
Adrenal cortical adenoma




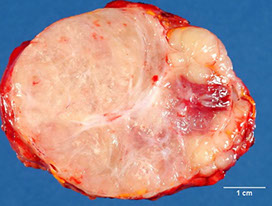
Black adenoma assoc c Cushing syndrome



Black adenoma
Diffusely brown-black ACA due to lipofuscin [1]
- usually lacks endocrine activity, though can be assoc c Cushings
- arises from the compact granulated cells of the zona reticularis and from the interface bwt this layer and the fasciculata zone
Micro: cords of cortical cells c brown pigmented granules (or can be in medulla), concentrates at corticomedullary junction




Adrenal Cortical Carcinoma (ACC)
Larger than adenomas (>200-300 g), usually unilateral
- assoc c Li-Fraumeni and Beckwith-Weidemann syndrome
- opposite adrenal may be atrophic (majority of these carcinomas are functional) and assoc c aldosteronism, Cushing syndrome, adrenal virilism, feminization
- also get general tumor symptoms such as weight loss, abdominal pain, back pain, night sweats, or anorexia
Criteria for malignancy:
- Radiology plays a major role in the diagnosis and staging of ACC
Tumor cytology is NOT sufficient to differentiate adrenal cortical adenoma and ACC since adrenal cortical adenoma may show bizarre atypia and ACC may be bland.
- metastasis or invasion into adjacent structures is sufficient to document malignancy
Gross: bulky yellow tumor, usually >20 cm, c necrosis, calcs, and bleeding
- breach of the adrenal capsule is an important gross characteristic, as well as invasion into adjacent structures
Micro: Tumor cytology is NOT sufficient to differentiate adrenal cortical adenoma and ACC since adrenal cortical adenoma may show bizarre atypia and ACC may be bland.
- may range from being cytologically bland to showing bizarre anaplasia.
- can be diffuse, hepatoid with sinusoids, or trabecular
- the presence of clear cells is regarded as a favorable feature
- has high mit rate (which are atypical), few clear cells, high-grade nuclei, necrosis, capsular invasion, vascular invasion
Variants with prognostic significance include oncocytic (best survival), myxoid, sarcomatoid (worst survival)
Modified Weiss scoring system
- Mitotic rate >5 / 50 hpfs = 2 pts
- Clear / vacuolated cells comprising <= 25% of tumor = 2 pts
- Atypical mitotic figures = 1 pt
- Necrosis = 1 pt
- Capsular invasion = 1 pt
Cumulative score >= 3 correlates with subsequent malignant behavior
- note that the Modified Weiss scoring system does not perform well as a determinant of malignancy with the oncocytic variant of ACC
IHC; (+) Melan-A, inhibin, calretinin, SF1 (new and most specific stain), pankeratin (usually weak, can be negative), SYN (variable, can be weak or negative), +/- HepPar1
- neg: S100 and HMB45, EMA, CHR
DDx:
- Adrenal cortical adenoma: modified Weiss system helps differentiate
- Pheochromocytoma: +CHR, GATA3, neg SF1, inhibin
- Hepatocellular carcinoma (also is HepPar1+, but is negative for SF1 and inhibin
- Chromophobe RCC: +EMA, CKIT, CK7 and E-cadherin, neg SF1
- Melanoma: +S100
- Neuroendocrine carcinoma: may met to adrenals, and is +CHR, but is neg for SF1
Px: Poor, often present late, 2-yr survival only 50%
- commonly mets to liver, bones, lungs, rp lymph nodes
Adrenal cortical carcinoma

Neuroblastic tumors
least to most diff: Neuroblastoma to Ganglioneuroblastoma to Ganglioneuroma
- derived from neural crest cells
- can have diarrhea from prod of VIP
Neuroblastoma
MC tumor of adrenal medulla in children (older than 7 yo) from neural crest cells made of immature neuroblasts, M>F, MC in adrenal medulla (about 2/5), though can occur anywhere alond adrenal sympathetic chain, usually bilateral
- may occur anywhere in the sympathetic chain
- assoc c raccoon eyes, rapid eye mvmts, paralysis
Sx: opsoclous-myoclonus syndrome in kiddos
Gross: large, bulky, soft white mass c areas of hemorrhage, necrosis and calcs
Micro: small round blue cells that stain c fibrillary stroma, neurofilament and have Homer-Wright pseudorosettes
- can classify as undifferentiated (no neuropil, no cytoplasm); poorly differentiated (some neutropil, <5% neuroblasts); differentiating (lots of neutopil, lots of neuroblasts (>5%)
- Homer-Wright pseudorosettes - surround delicate red neuropil
Cyto: Neuroblasts: uniform, small round blue cells c red fibrillary cytoplasm, maybe Homer-Wright rossettes
IHC: (+) NSE, CD56/57, synapto, CHR, SYN, GFAP, S100 (septae)
- negative EMA, ck, vimentin, HMB45, WT1, CD45/99, myogenic stuff
Genetics: can be familial (PHOX2B or ALK mutations) or sporadic (ALK and lots of mutations in N-myc oncogene assoc c poor px)
- ATRX mutations common, also in Rac/Rho pathways
- Trk gene expression (encodes nerve growth factor for neuroblast differentiation), assoc c good px
- hyperdiploidy and near-triploidy assoc c good px
Dx: HVA (breakdown product of dopamine) in urine, VMA can be seen as intracytoplasmic granules by EM
Tx: anywhere from observation to excision to radio-chemo immunotx
Px: many different classifications and factors, age and stage most important determinants
- though usually aggressive and highly malignant
- can have different types of mets: Pepper-type (massive liver mets), Hutchinson-type (skull and orbit involvement c exopthalmos), micromets to skin, bone marrow, or liver
- spontaneous regression or differentation of tumor cells to ganglioneuroblastoma or mature ganlgioneuroma can happen
Neuroblastoma - bulky but relatively well-circumscribed solid tumor
Neoroblastoma of adrenal gland
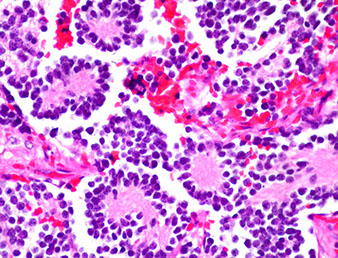
Neuroblastoma


Ganglioneuroblastoma
mix of immature neuroblasts and ganglion cells, stroma has lots of Swann cells
- usually b4 4 yo, M=F, 4/5 in adrenals, neural crest origin
- present at advanced stage
- labs also show catecholamine metabolites in urine (HMA, VMA)
Grossly, more homogenous and mature appearing than neuroblastoma
Micro: Neuroblasts can have Homer Wright pseudorosettes, minimal cytoplasm, salt n peppa chromatin
- Ganglion cells have lots of red cytoplasm, more well defined, big nuclei
- small round blue cell (neuroblastemal) component is neuroblastoma
Cyto: can see neuroblasts and ganglion cells
IHC: similar to neuroblastoma
Genes: heterogenous, not well understood
ganglioneuroblastoma - neuroblasts
ganglioneuroblastoma - ganglion cells

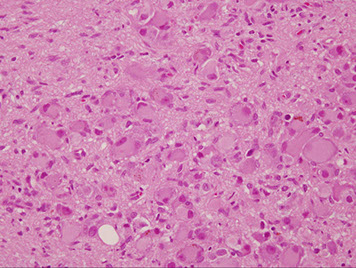
Ganglioneuroma
Neural crest cell derivative, usually in kiddos > 7 yo, MC in posterior mediastinum, mix of ganglion cells and Schwann stromal cells
- familial disorders assoc c Turners and MEN2
Micro: mix of ganglion cells (can be immature, must state so if dont look completely mature), Schwann cells in intersecting fascicles c loose myxoid stroma
- no sig atypia or necrosis
Cyto: biphasic, large ganglion cells c spindly Schwann cells
IHC: (+) S100, synapto, NF protein, type IV collagen
- negative EMA, CKs, HMB45, WT1, CD45/99
Genes: usually normal
Tx: take out adrenal

ganglioneuroma
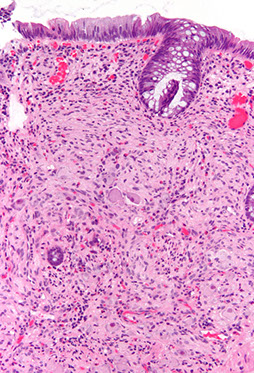
Myelolipoma
B9 mesenchymal tumor of adrenal cortex that is made of hematopoietic precursor cells (looks like extramedullary hematopoiesis [EMH]) and fat
- usually asymptomatic and is incidental finding on CT or at autopsy
- is a type of "incidentaloma" as b9 mesenchymal tumor of cortex, no clinical significance
- not assoc c hematologic disorders; nonfunctioning
- may be due to metaplastic change in reticuloendothelial cells in response to stimuli including necrosis, infection or stress
Gross: sharply circumscribed, unencapsulated and pale yellow
Micro: fat cells mixed c hematopoietic cells (megakaryocytes, erythroid and myeloid cells)
Tx: excision if symptomatic
Paraganglioma
- see Nervous System Tumors
References
1. Osler notes
2. PathologyOutlines.com